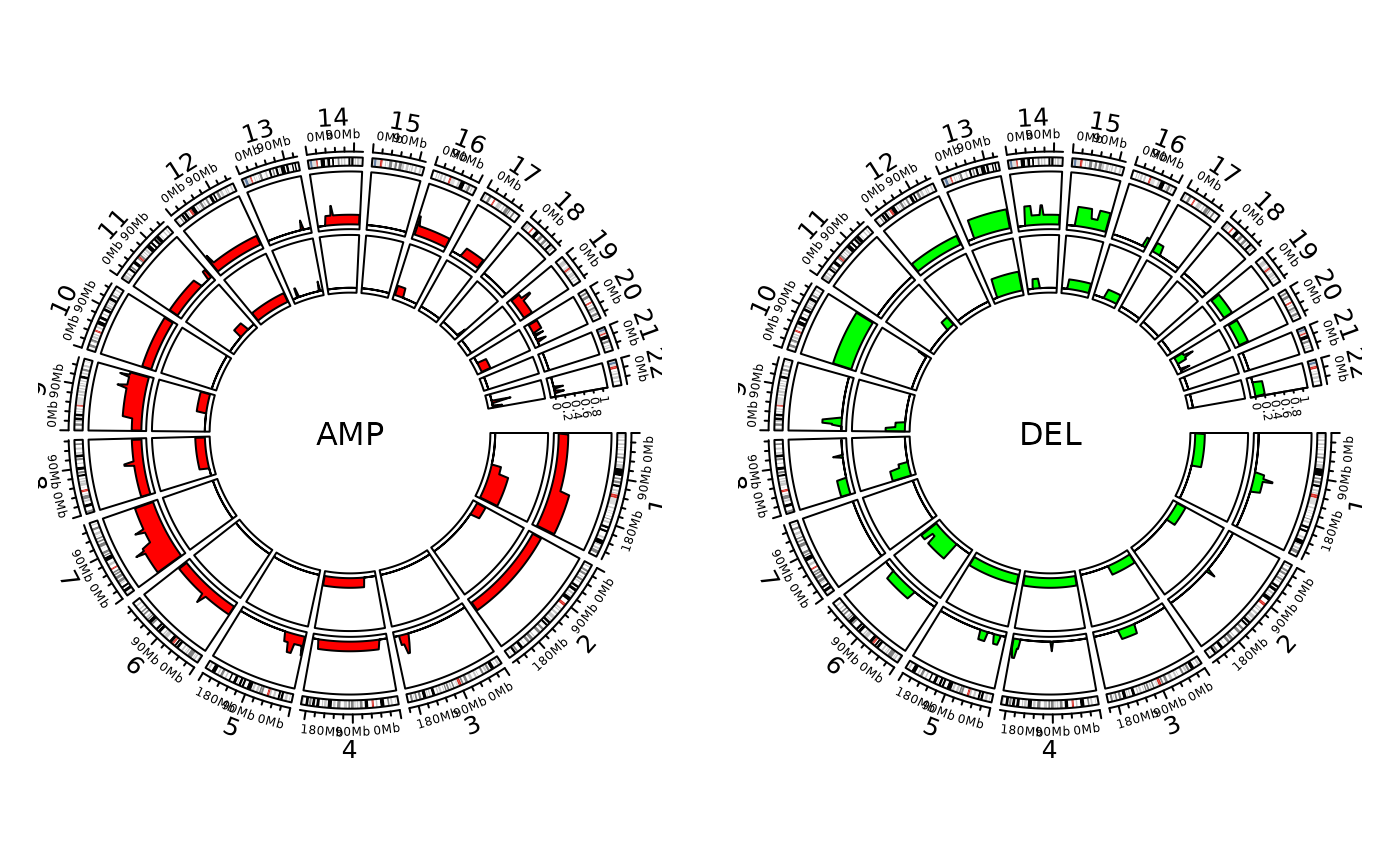
Show Copy Number Variation Frequency Profile with Circos
Source:R/show_cn_freq_circos.R
show_cn_freq_circos.RdShow Copy Number Variation Frequency Profile with Circos
Arguments
- data
a
CopyNumberobject or a data.frame containing at least 'chromosome', 'start', 'end', 'segVal', 'sample' these columns.- groups
a named list or a column name for specifying groups.
- cutoff
copy number value cutoff for splitting data into AMP and DEL. The values equal to cutoff are discarded. Default is
2, you can also set a length-2 vector, e.g.c(2, 2).- resolution_factor
an integer to control the resolution. When it is
1(default), compute frequency in each cytoband. When it is2, use compute frequency in each half cytoband.- title
length-2 titles for AMP and DEL.
- chrs
chromosomes start with 'chr'.
- genome_build
genome build version, used when
datais adata.frame, should be 'hg19' or 'hg38'.- cols
length-2 colors for AMP and DEL.
- plot_ideogram
default is
TRUE, show ideogram.- track_height
track height in
mmunit.- ideogram_height
ideogram height in
mmunit.- ...
other parameters passing to circlize::circos.genomicLines.
Value
Nothing.
Examples
# \donttest{
load(system.file("extdata", "toy_copynumber.RData",
package = "sigminer", mustWork = TRUE
))
show_cn_freq_circos(cn)
#> Plotting AMP
#> Plotting group 1
#> Plotting DEL
#> Plotting group 1
 ss <- unique(cn@data$sample)
show_cn_freq_circos(cn, groups = list(a = ss[1:5], b = ss[6:10]), cols = c("red", "green"))
#> Plotting AMP
#> Plotting group a
#> Plotting group b
#> Plotting DEL
#> Plotting group a
#> Plotting group b
ss <- unique(cn@data$sample)
show_cn_freq_circos(cn, groups = list(a = ss[1:5], b = ss[6:10]), cols = c("red", "green"))
#> Plotting AMP
#> Plotting group a
#> Plotting group b
#> Plotting DEL
#> Plotting group a
#> Plotting group b
 # }
# }