Attaching SeuratObject# load dataset
data("ifnb")学习资料:GitHub 地址
这个教程在运行到 predict_ligand_activities() 就出现了问题。后面就走不下去了。
predict_ligand_activities(geneset = geneset_oi, background_expressed_genes = background_expressed_genes, ligand_target_matrix = ligand_target_matrix, potential_
ligands = potential_ligands)
Error in evaluate_target_prediction(setting, ligand_target_matrix, ligands_position) :
all genes have same response我自己也懒得折腾了,本身就是想简单学习了解流程,走不通就不走了。如果有读者看到这个文章,感兴趣可以钻研下。
我查了下,Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data 显示CellChat、CellPhoneDB和SingleCellSignalR,对数据和资源中的噪声都相当稳健。因而NicheNet并不在推荐范围内。
不过也提到:
Methods that additionally infer intracellular processes, such as NicheNet19, Cytotalk22, and SoptSC20 are not directly comparable but instead provide complementary analyses.
说明目前看,还是有一些特定的分析有可取之处。
我们将使用SeuratData软件包提供的PBMC数据集。该数据集包含两个PBMC样本:经过IFN Beta处理的刺激样本和对照样本。
对于细胞交互分析,我们将使用NicheNet,它需要一些精心策划的数据集。虽然这些数据集可以在R环境内下载,但对我来说下载速度太慢了。这些文件可以从Zenodo下(https://zenodo.org/record/3260758)载:gr_network.rds、ligand_target_matrix.rds、lr_network.rds和weighted_networks.rds。
请将这些数据集放到你的工作目录中。
本文参考 Seurat 数据整合文档进行分析的输入数据准备,假设你对数据预处理部分已经很熟悉。
安装所需要的包。
BiocManager::install("limma")
BiocManager::install("glmGamPoi")
# install.packages("devtools")
install.packages("https://seurat.nygenome.org/src/contrib/ifnb.SeuratData_3.0.0.tar.gz", repos = NULL, type = "source")
devtools::install_github("saeyslab/nichenetr")
# 可以下载之后安装,避免安装不上
# https://github.com/saeyslab/nichenetr
# unzip("~/Downloads/nichenetr-master.zip")
# 缺什么装什么
# BiocManager::install(c("fdrtool", "DiagrammeR", "mlrMBO", "parallelMap", "emoa", "DiceKriging"))
# install.packages("nichenetr-master", repos = NULL, type = "source")
install.packages("tidyverse")加载包:
第一步是从SeuratData软件包中读取数据。输入数据根据样本进行拆分,并分别应用SCtranform。请注意,这取代了NormalizeData、ScaleData和FindVariableFeatures的步骤。SCTransform是一种专门设计用于单细胞UMI计数数据的统计方法。它克服了之前批量设计的归一化方法中一些过度拟合的限制。有关其优势的更多详细信息可以在SCtranform的论文中找到。请注意,我们没有应用任何质控步骤。这是因为数据已经预先清洁,因此无需重复这些步骤。
ifnb.list <- SplitObject(ifnb, split.by = "stim")
ifnb.list <- lapply(X = ifnb.list, FUN = SCTransform, method = "glmGamPoi")Calculating cell attributes from input UMI matrix: log_umiVariance stabilizing transformation of count matrix of size 12747 by 6548Model formula is y ~ log_umiGet Negative Binomial regression parameters per geneUsing 2000 genes, 5000 cells
|
| | 0%
|
|================== | 25%
|
|=================================== | 50%
|
|==================================================== | 75%
|
|======================================================================| 100%Found 95 outliers - those will be ignored in fitting/regularization stepSecond step: Get residuals using fitted parameters for 12747 genes
|
| | 0%
|
|=== | 4%
|
|===== | 8%
|
|======== | 12%
|
|=========== | 15%
|
|============= | 19%
|
|================ | 23%
|
|=================== | 27%
|
|====================== | 31%
|
|======================== | 35%
|
|=========================== | 38%
|
|============================== | 42%
|
|================================ | 46%
|
|=================================== | 50%
|
|====================================== | 54%
|
|======================================== | 58%
|
|=========================================== | 62%
|
|============================================== | 65%
|
|================================================ | 69%
|
|=================================================== | 73%
|
|====================================================== | 77%
|
|========================================================= | 81%
|
|=========================================================== | 85%
|
|============================================================== | 88%
|
|================================================================= | 92%
|
|=================================================================== | 96%
|
|======================================================================| 100%Computing corrected count matrix for 12747 genes
|
| | 0%
|
|=== | 4%
|
|===== | 8%
|
|======== | 12%
|
|=========== | 15%
|
|============= | 19%
|
|================ | 23%
|
|=================== | 27%
|
|====================== | 31%
|
|======================== | 35%
|
|=========================== | 38%
|
|============================== | 42%
|
|================================ | 46%
|
|=================================== | 50%
|
|====================================== | 54%
|
|======================================== | 58%
|
|=========================================== | 62%
|
|============================================== | 65%
|
|================================================ | 69%
|
|=================================================== | 73%
|
|====================================================== | 77%
|
|========================================================= | 81%
|
|=========================================================== | 85%
|
|============================================================== | 88%
|
|================================================================= | 92%
|
|=================================================================== | 96%
|
|======================================================================| 100%Calculating gene attributesWall clock passed: Time difference of 31.92474 secsDetermine variable featuresPlace corrected count matrix in counts slotCentering data matrixSet default assay to SCTCalculating cell attributes from input UMI matrix: log_umiVariance stabilizing transformation of count matrix of size 12658 by 7451Model formula is y ~ log_umiGet Negative Binomial regression parameters per geneUsing 2000 genes, 5000 cells
|
| | 0%
|
|================== | 25%
|
|=================================== | 50%
|
|==================================================== | 75%
|
|======================================================================| 100%Found 101 outliers - those will be ignored in fitting/regularization stepSecond step: Get residuals using fitted parameters for 12658 genes
|
| | 0%
|
|=== | 4%
|
|===== | 8%
|
|======== | 12%
|
|=========== | 15%
|
|============= | 19%
|
|================ | 23%
|
|=================== | 27%
|
|====================== | 31%
|
|======================== | 35%
|
|=========================== | 38%
|
|============================== | 42%
|
|================================ | 46%
|
|=================================== | 50%
|
|====================================== | 54%
|
|======================================== | 58%
|
|=========================================== | 62%
|
|============================================== | 65%
|
|================================================ | 69%
|
|=================================================== | 73%
|
|====================================================== | 77%
|
|========================================================= | 81%
|
|=========================================================== | 85%
|
|============================================================== | 88%
|
|================================================================= | 92%
|
|=================================================================== | 96%
|
|======================================================================| 100%Computing corrected count matrix for 12658 genes
|
| | 0%
|
|=== | 4%
|
|===== | 8%
|
|======== | 12%
|
|=========== | 15%
|
|============= | 19%
|
|================ | 23%
|
|=================== | 27%
|
|====================== | 31%
|
|======================== | 35%
|
|=========================== | 38%
|
|============================== | 42%
|
|================================ | 46%
|
|=================================== | 50%
|
|====================================== | 54%
|
|======================================== | 58%
|
|=========================================== | 62%
|
|============================================== | 65%
|
|================================================ | 69%
|
|=================================================== | 73%
|
|====================================================== | 77%
|
|========================================================= | 81%
|
|=========================================================== | 85%
|
|============================================================== | 88%
|
|================================================================= | 92%
|
|=================================================================== | 96%
|
|======================================================================| 100%Calculating gene attributesWall clock passed: Time difference of 30.21689 secsDetermine variable featuresPlace corrected count matrix in counts slotCentering data matrixSet default assay to SCTfeatures <- SelectIntegrationFeatures(object.list = ifnb.list, nfeatures = 3000)
ifnb.list <- PrepSCTIntegration(object.list = ifnb.list, anchor.features = features)
ifnb.list <- lapply(X = ifnb.list, FUN = RunPCA, features = features)PC_ 1
Positive: FTL, TIMP1, IL8, CCL2, FTH1, C15orf48, S100A8, SOD2, ACTB, S100A9
LYZ, FCER1G, TYROBP, S100A4, HLA-DRA, CD63, CD14, S100A11, LGALS3, ANXA5
LGALS1, CXCL3, S100A10, IL1B, CTSL, PLAUR, CTSB, TYMP, S100A6, CST3
Negative: RPL3, RPS18, RPS6, LTB, RPL13, CCR7, GIMAP7, RPL13A, PTMA, CXCR4
RPSA, RPS2, RPL21, CD3D, RPS5, LDHB, NPM1, RPS4X, CCL5, RPS3
TMEM66, RPS14, RPS27, PABPC1, RPL10A, RPS15A, RPL5, RPL32, RPS19, RPL34
PC_ 2
Positive: CCL2, CCL5, GNLY, S100A8, NKG7, FTL, S100A9, GZMB, GIMAP7, CD7
CD3D, CD14, FTH1, IL8, CXCL3, CD2, FGFBP2, CCL7, RGCC, RARRES3
GZMH, APOBEC3G, TMEM66, CLIC3, ITM2A, CTSL, LTB, IL32, LCK, GIMAP5
Negative: HLA-DRA, CD74, HLA-DQA1, HLA-DPA1, HLA-DRB1, HLA-DPB1, HLA-DQB1, CD83, TXN, CCR7
CCL22, HLA-DMA, IDO1, ACTB, HERPUD1, LYZ, SYNGR2, CD79A, MIR155HG, BIRC3
PKIB, RAB9A, SERPINB1, RAMP1, GPR183, CST3, FABP5, HLA-DRB5, TVP23A, ID2
PC_ 3
Positive: RPS18, RPL13, LTB, RPS6, FTL, RPL3, RPL32, RPL10, RPS2, PABPC1
RPL34, RPL13A, RPS14, RPS4X, RPL21, S100A8, CCL2, RPS12, RPL11, RPL7
RPS3A, RPL18A, LDHB, SELL, RPS15A, RPS3, RPS8, S100A9, RPS5, RPS27A
Negative: GNLY, NKG7, CCL5, GZMB, CST7, APOBEC3G, FGFBP2, CLIC3, GZMH, KLRD1
GZMA, CTSW, PRF1, HOPX, KLRC1, CHST12, TNFRSF18, CCL4, VMO1, FCGR3A
DUSP2, LDHA, CXCR3, C1orf21, ID2, MATK, SPON2, SH2D1B, RARRES3, APMAP
PC_ 4
Positive: HSPB1, HSP90AB1, HSPE1, CACYBP, CCL2, HSPA8, CREM, HSPD1, SRSF7, HSPH1
HSPA1A, FTL, CD69, UBB, GADD45B, UBC, SRSF2, ZFAND2A, HSP90AA1, IL8
NOP58, DDIT4, NR4A2, CHORDC1, H3F3B, BTG1, RSRC2, DNAJB1, SNHG15, YPEL5
Negative: VMO1, FCGR3A, TIMP1, MS4A7, RPL3, FAM26F, TNFSF10, RPS4X, RPS2, RPS19
RPL10, RPS18, PLAC8, CXCL16, CXCL10, HLA-DPA1, LST1, HLA-DPB1, RPL13, TNFSF13B
MS4A4A, RPS14, IFITM3, HN1, AIF1, GBP5, RPS6, CCL5, CD74, GBP1
PC_ 5
Positive: CCL2, FTL, GNLY, CCL5, NKG7, IL8, GZMB, S100A8, S100A9, LYZ
HLA-DRA, CST7, CD74, HLA-DQA1, CXCL3, TXN, FGFBP2, CD63, GAPDH, CLIC3
IL1B, HLA-DPB1, HLA-DRB1, VIM, RPL3, GZMH, CCL7, APOBEC3G, C15orf48, HLA-DQB1
Negative: VMO1, FCGR3A, MS4A7, TIMP1, FAM26F, TNFSF10, CXCL10, HSPB1, IFITM3, GBP1
AIF1, PLAC8, MS4A4A, LST1, GBP5, CXCL16, HN1, SAT1, ATP1B3, OAS1
ISG15, C3AR1, WARS, CTSC, PPM1N, RGS2, TNFSF13B, SOD2, IFIT3, FGL2 PC_ 1
Positive: FTL, CXCL10, CCL2, CCL8, APOBEC3A, SOD2, TIMP1, C15orf48, CCL7, APOBEC3B
ISG15, IDO1, CCL3, IFITM3, CXCL11, LYZ, TYROBP, CCL4, IFI27, FCER1G
FTH1, CTSL, SAT1, TYMP, IL1RN, LGALS3, S100A11, CTSC, CD63, LGALS1
Negative: RPL3, RPS6, RPS18, RPL13, RPL21, RPL7, RPL13A, CCR7, RPL10A, RPS4X
RPS2, GIMAP7, RPS3, RPS3A, RPS14, RPS27A, PTMA, RPL10, RPS15A, RPL34
RPS5, RPL32, PABPC1, RPL5, CXCR4, RPS27, RPL31, LTB, MALAT1, RPL14
PC_ 2
Positive: CCL2, CCL8, CCL7, RPS18, RPL13, RPS6, FTL, RPL3, RPL32, SELL
RPS14, RPL34, LTB, RPL7, RPL13A, RPL21, RPL10, GIMAP7, PABPC1, RPS2
RPS4X, APOBEC3B, RPL10A, RPS15A, RPS3A, LDHB, S100A9, RPS12, RPL35A, RPL31
Negative: GNLY, GZMB, NKG7, CCL5, PRF1, CD74, CLIC3, CST7, GZMA, HLA-DRA
APOBEC3G, GZMH, HLA-DPB1, HLA-DPA1, KLRD1, CTSW, TXN, FCGR3A, HLA-DQA1, VMO1
HLA-DRB1, FASLG, ID2, FGFBP2, CD83, KLRC1, MS4A7, HLA-DQB1, RARRES3, TIMP1
PC_ 3
Positive: CD74, HLA-DRA, HLA-DPB1, CXCL10, HLA-DPA1, HLA-DQA1, HLA-DRB1, TXN, TIMP1, CD83
VMO1, FCGR3A, CCR7, HLA-DQB1, MS4A7, MARCKSL1, IRF8, CST3, BIRC3, SYNGR2
MS4A4A, SERPINB1, CD79A, CXCL16, CD86, MIR155HG, HLA-DMA, CCL22, ACTB, FAM26F
Negative: GNLY, CCL2, CCL8, NKG7, CCL5, GZMB, CCL7, PRF1, GZMA, GZMH
CLIC3, KLRD1, CTSW, RARRES3, S100A9, ALOX5AP, FASLG, APOBEC3G, FGFBP2, CTSL
CD7, CCL4, KLRC1, CST7, S100A8, ANXA1, CCL3, CD247, IL32, LGALS3
PC_ 4
Positive: FCGR3A, VMO1, MS4A7, TIMP1, MS4A4A, CXCL16, FAM26F, PLAC8, TNFSF10, TNFAIP6
C3AR1, LST1, FTH1, CXCL10, TYROBP, CTSC, AIF1, GBP5, PPM1N, CFD
MT2A, CDKN1C, SERPINA1, IFITM3, SLC31A2, HN1, PLAUR, FCER1G, FCGR3B, EDN1
Negative: CCL2, CCL8, CD74, HLA-DRA, TXN, HLA-DQA1, CCL7, HLA-DPB1, FTL, HLA-DRB1
LYZ, MIR155HG, HLA-DQB1, CD83, IRF8, FABP5, CCR7, MARCKSL1, BIRC3, APOBEC3B
TSPAN13, HERPUD1, HSPE1, HSPD1, HLA-DPA1, ID3, PMAIP1, HSP90AB1, CD79A, NME1
PC_ 5
Positive: CD74, HLA-DPB1, HLA-DRA, RPL3, TXN, RPS18, RPS4X, MARCKSL1, HLA-DPA1, RPL10
RPL13, LYZ, HLA-DRB1, FTL, RPS2, GNLY, RPS14, GZMB, CCL5, RPS3
RPL34, NKG7, RPL11, RPL13A, CST3, RPL32, RPS6, RPL21, HLA-DQA1, LSP1
Negative: CCL4, CCL3, CD69, SRSF7, HSPE1, HSP90AB1, CACYBP, HSPA8, HSPD1, SRSF2
DDIT4, CREM, GADD45B, UBC, NOP58, UBB, HSPH1, TCP1, JUNB, SOD1
SAT1, CLK1, RSRC2, BTG1, DNAJB6, NR4A2, MYC, IRF1, DNAJB1, BATF 接下来,我们将两个数据集进行整合。整合依赖于在两个数据集中都存在的高变量特征,并且分为两个步骤。第一步FindIntegrationAnchors定义锚点或细胞对(每个样本中各选择一个细胞),这些细胞非常相似,因此可以有信心地将它们分配到相同的簇(细胞类型和状态)。第二步IntegrateData使用所定义的锚点对齐完整的数据集。有关整合方法的更多详细信息和自原始Seurat论文以来的改进可以在他们最近的出版物中找到。
immune.anchors <- FindIntegrationAnchors(object.list = ifnb.list, normalization.method = "SCT", anchor.features = features, dims = 1:30, reduction = "rpca", k.anchor = 20)Computing within dataset neighborhoodsFinding all pairwise anchorsProjecting new data onto SVD
Projecting new data onto SVDFinding neighborhoodsFinding anchors Found 32565 anchorsimmune.combined.sct <- IntegrateData(anchorset = immune.anchors, normalization.method = "SCT", dims = 1:30)Merging dataset 1 into 2Extracting anchors for merged samplesFinding integration vectorsFinding integration vector weightsIntegrating data整合后的对象包含经过批次效应校正的值,以及原始计数值作为单独的测定。
immune.combined.sct <- RunPCA(immune.combined.sct, verbose = FALSE)
immune.combined.sct <- RunUMAP(immune.combined.sct, reduction = "pca", dims = 1:30)Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
This message will be shown once per session15:39:33 UMAP embedding parameters a = 0.9922 b = 1.11215:39:33 Read 13999 rows and found 30 numeric columns15:39:33 Using Annoy for neighbor search, n_neighbors = 3015:39:33 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
15:39:35 Writing NN index file to temp file /var/folders/bj/nw1w4g1j37ddpgb6zmh3sfh80000gn/T//RtmpF254ot/file1575b69d2520a
15:39:35 Searching Annoy index using 1 thread, search_k = 3000
15:39:38 Annoy recall = 100%
15:39:39 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
15:39:40 Initializing from normalized Laplacian + noise (using irlba)
15:39:41 Commencing optimization for 200 epochs, with 600044 positive edges
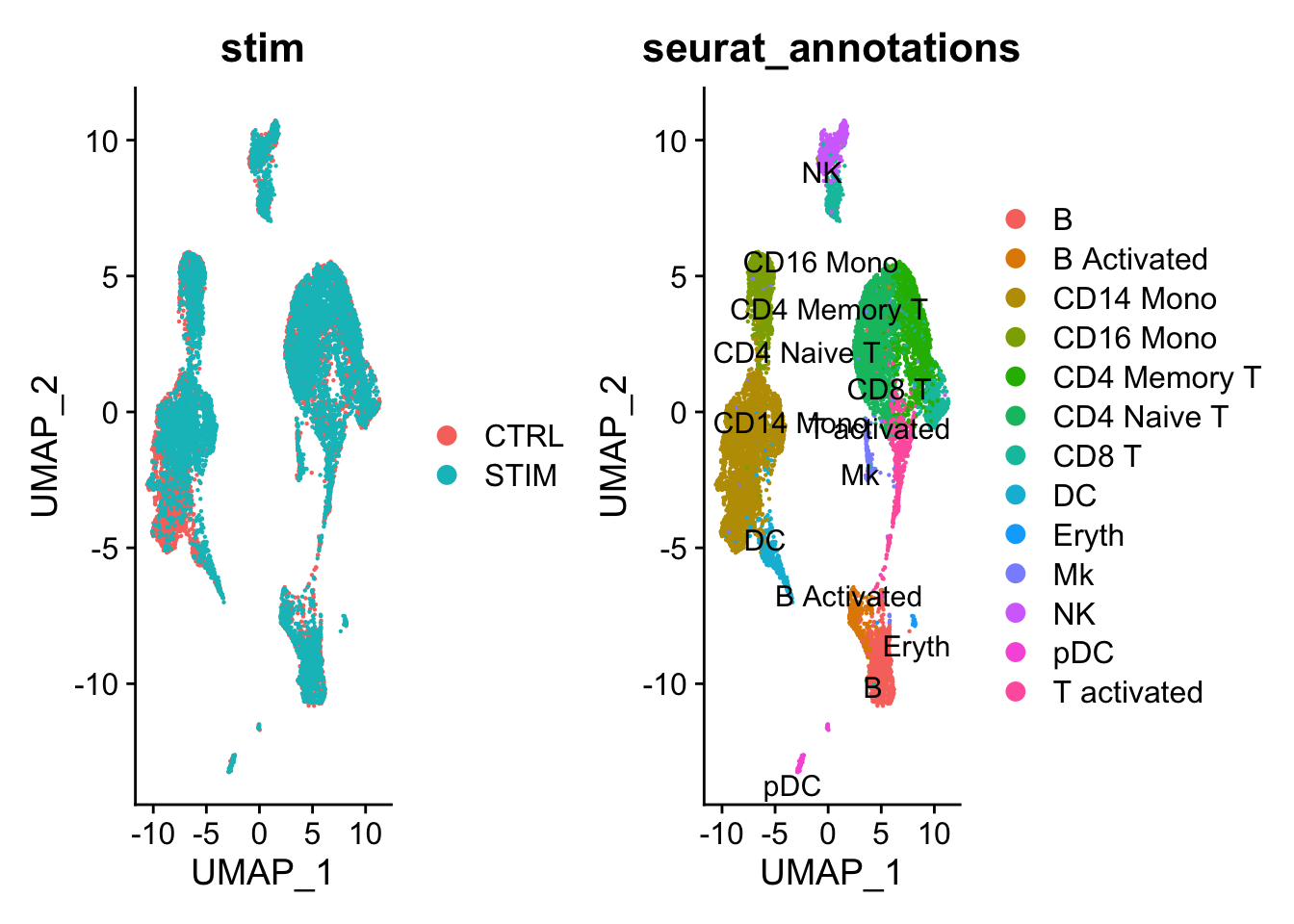
15:39:49 Optimization finished可视化:
NicheNet旨在通过将特定簇的单细胞表达数据与关于配体-受体对和目标下游的基因调控网络的先验知识相结合,预测配体和目标细胞之间的相互作用连接。特别地,它旨在定义能够最好解释目标簇中观察到的差异表达的配体。
我们首先加载必要的R软件包。⚠️在我加载了NicheNet软件包后,部分Seurat整合命令未能成功执行。两个软件包之间可能存在一些不兼容性。
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.2 ✔ readr 2.1.4
✔ forcats 1.0.0 ✔ stringr 1.5.0
✔ ggplot2 3.4.2 ✔ tibble 3.2.1
✔ lubridate 1.9.2 ✔ tidyr 1.3.0
✔ purrr 1.0.1
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors加载数据集:
ligand_target_matrix = readRDS("/Users/wsx/Library/CloudStorage/OneDrive-shanghaitech.edu.cn/Public/data/ligand_target_matrix.rds")
ligand_target_matrix[1:5,1:5] CXCL1 CXCL2 CXCL3 CXCL5 PPBP
A1BG 3.534343e-04 4.041324e-04 3.729920e-04 3.080640e-04 2.628388e-04
A1BG-AS1 1.650894e-04 1.509213e-04 1.583594e-04 1.317253e-04 1.231819e-04
A1CF 5.787175e-04 4.596295e-04 3.895907e-04 3.293275e-04 3.211944e-04
A2M 6.027058e-04 5.996617e-04 5.164365e-04 4.517236e-04 4.590521e-04
A2M-AS1 8.898724e-05 8.243341e-05 7.484018e-05 4.912514e-05 5.120439e-05lr_network = readRDS("/Users/wsx/Library/CloudStorage/OneDrive-shanghaitech.edu.cn/Public/data/lr_network.rds")
head(lr_network)# A tibble: 6 × 4
from to source database
<chr> <chr> <chr> <chr>
1 CXCL1 CXCR2 kegg_cytokines kegg
2 CXCL2 CXCR2 kegg_cytokines kegg
3 CXCL3 CXCR2 kegg_cytokines kegg
4 CXCL5 CXCR2 kegg_cytokines kegg
5 PPBP CXCR2 kegg_cytokines kegg
6 CXCL6 CXCR2 kegg_cytokines kegg weighted_networks = readRDS("/Users/wsx/Library/CloudStorage/OneDrive-shanghaitech.edu.cn/Public/data/weighted_networks.rds")
weighted_networks_lr = weighted_networks$lr_sig %>% inner_join(lr_network %>% distinct(from,to), by = c("from","to"))
head(weighted_networks$lr_sig) # interactions and their weights in the ligand-receptor + signaling network# A tibble: 6 × 3
from to weight
<chr> <chr> <dbl>
1 A1BG ABCC6 0.422
2 A1BG ACE2 0.101
3 A1BG ADAM10 0.0970
4 A1BG AGO1 0.0525
5 A1BG AKT1 0.0855
6 A1BG ANXA7 0.457 Idents(immune.combined.sct) <-immune.combined.sct$seurat_annotations
DefaultAssay(immune.combined.sct) <- "integrated"在本文中,我们依赖于数据提供的筛选细胞注释。 我们将重点放在CD8 T细胞作为接收细胞,并寻找pDC、CD14 Mono、CD16 Mono、NK、B、DC、B Activated群集中的发送器配体。
我们定义接收细胞和发送器细胞群体。此外,我们还识别在接收细胞中表达的基因,这些基因也是我们知识库的一部分。
💡如果某个基因在数据库中没有被知道由任何配体调控,该方法将无法在其预测中使用它。
receiver = "CD8 T"
expressed_genes_receiver = get_expressed_genes(receiver, immune.combined.sct, pct = 0.10, assay_oi="RNA")
background_expressed_genes = expressed_genes_receiver %>% .[. %in% rownames(ligand_target_matrix)]
sender_celltypes = c("pDC", "CD14 Mono" , "CD16 Mono", "NK", "B", "DC", "B Activated")
list_expressed_genes_sender = sender_celltypes %>% unique() %>% lapply(get_expressed_genes, immune.combined.sct, 0.10, assay_oi="RNA")
expressed_genes_sender = list_expressed_genes_sender %>% unlist() %>% unique()定义接收细胞中相对于样本处理的差异基因。
seurat_obj_receiver= subset(immune.combined.sct, idents = receiver)
seurat_obj_receiver = SetIdent(immune.combined.sct, value = seurat_obj_receiver[["orig.ident"]])
condition_oi = "IMMUNE_STIM"
condition_reference = "IMMUNE_CTRL"
DE_table_receiver = FindMarkers(object = seurat_obj_receiver, ident.1 = condition_oi, ident.2 = condition_reference, min.pct = 0.10, assay="RNA") %>% rownames_to_column("gene")
geneset_oi = DE_table_receiver %>% filter(p_val <= 0.05 & abs(avg_log2FC) >= 0.25) %>% pull(gene)
geneset_oi = geneset_oi %>% .[. %in% rownames(ligand_target_matrix)]定义在各自群集中表达的配体和受体基因。潜在的配体集被缩小为在接收细胞群集中表达的具有靶标的配体。
ligands = lr_network %>% pull(from) %>% unique()
receptors = lr_network %>% pull(to) %>% unique()
expressed_ligands = intersect(ligands,expressed_genes_sender)
expressed_receptors = intersect(receptors,expressed_genes_receiver)
potential_ligands = lr_network %>% filter(from %in% expressed_ligands & to %in% expressed_receptors) %>% pull(from) %>% unique()NicheNet的配体活性分析根据接收细胞中存在的表达靶标基因的情况对配体进行排名。
# ligand_activities = predict_ligand_activities(geneset = geneset_oi, background_expressed_genes = background_expressed_genes, ligand_target_matrix = ligand_target_matrix, potential_ligands = potential_ligands)
# ligand_activities = ligand_activities %>% arrange(-pearson) %>% mutate(rank = rank(desc(pearson)))
# ligand_activities