本系统提供了一个使用四种方法(CIRIquant、Circexplorer2、find_circ和circRNA_finder)从转录组双端测序的FASTQ文件中轻松且可重复地检测环状RNA(circRNA)的计算流程。
功能特性
- 多方法鉴定和整合
- 并行计算支持
- 断点续跑
- 结果整合和规整化输出
- 日志记录
准备工作
操作系统环境
本系统需要运行在 Linux 系统环境中(不限发行版本,测试系统包括 Ubuntu、CentOS)。 需要用户知道如何操作 Linux 系统。
步骤1. 安装所需的conda环境
(可选)创建一个名为’circrna’的独立Linux帐户,用于部署和运行循环RNA识别流程。
安装miniconda3到默认路径,即
~/miniconda3。 如果按照上述推荐设置,conda应该可在/home/circrna/miniconda3处使用。使用
conda install -n base --override-channels -c conda-forge mamba 'python_abi=*=*cp*'将mamba安装到base环境中。使用以下命令安装just:
curl --proto '=https' --tlsv1.2 -sSf https://just.systems/install.sh | bash -s -- --to ~/bin。请将~/bin添加到您的$PATH中。 您可以将~/bin更改为任何位置,但在进入终端时需要使just可用。安装rush并将其路径添加到
$PATH,类似于just。(可选)如果需要,设置conda和pypi(pip)的注册表。例如,如果您在中国,我建议使用以下链接进行设置:https://mirrors.tuna.tsinghua.edu.cn/help/anaconda/ 和 https://mirrors.tuna.tsinghua.edu.cn/help/pypi/。
使用本软著提供的代码,并切换到代码目录。
按顺序安装conda环境。
cd circrna-pipeline cd CIRIquant just install cd ../FindCirc just install cd ../Circexplorer2 just install cd ../circRNA_finder just install
请确保已创建所有conda环境并安装了所需的软件。
步骤2. 准备比对索引、参考文件和配置文件
要运行流程,需要准备好一些参考数据文件。
准备基因组fasta文件和gtf文件。我们使用
GRCh38.primary_assembly.genome.fa和gencode.v34.annotation.gtf。对于Circexplorer2,您需要使用
Circexplorer2环境中的fetch_ucsc.py脚本下载参考文件hg38_ref_all.txt(应与您的参考基因组相对应)。准备比对索引,config_zhou.sh中记录了命令和配置,截图如下。
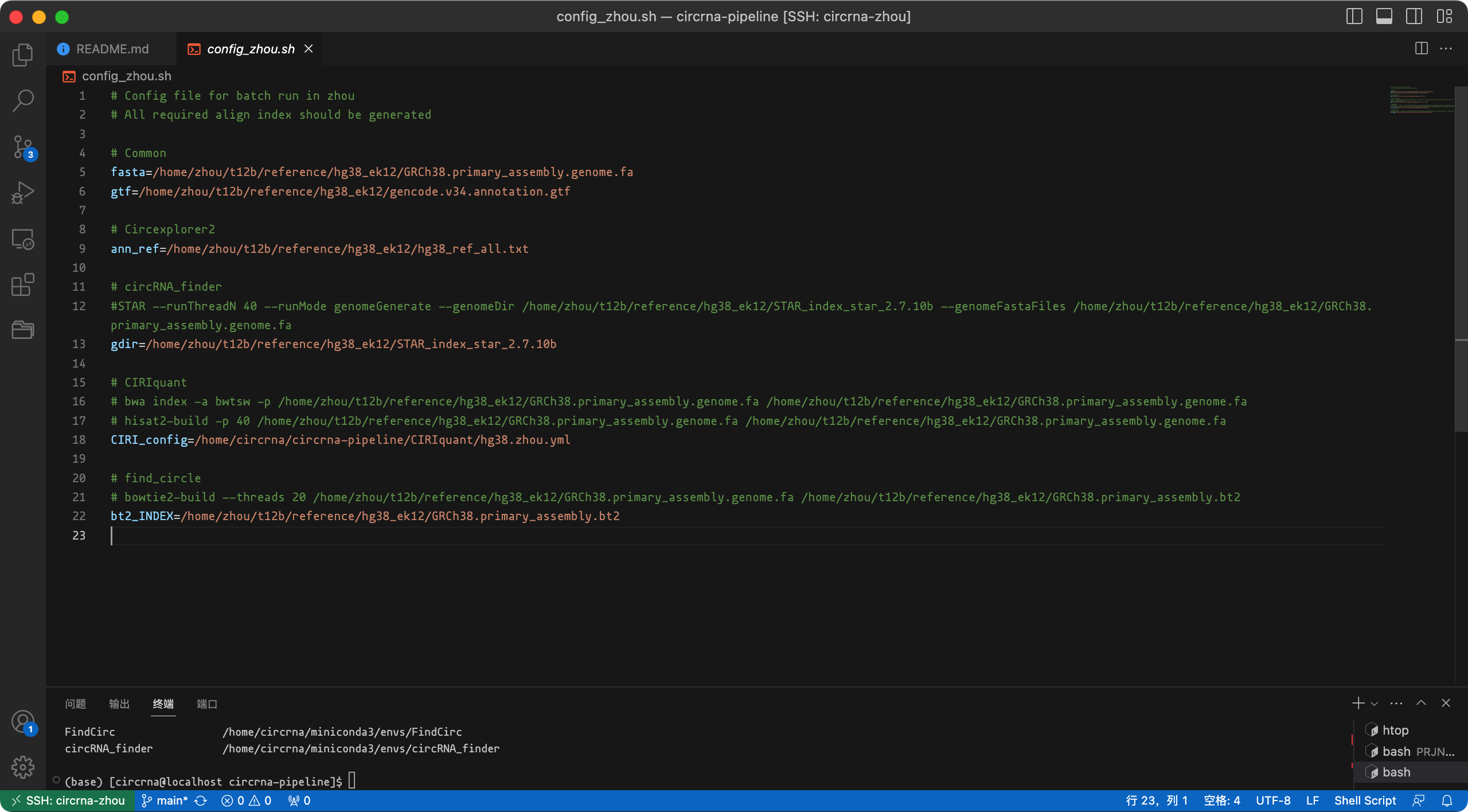
image-20230903160258992 请注意,在运行索引命令之前,您需要激活相应的环境。
例如,为CIRIquant准备索引。
sh source activate CIRIquant bwa index -a bwtsw -p /path/to/GRCh38.primary_assembly.genome.fa /path/to/GRCh38.primary_assembly.genome.fa hisat2-build -p 40 /path/to/GRCh38.primary_assembly.genome.fa /path/to/GRCh38.primary_assembly.genome.fa对于CIRIquant,需要一个
yml文件来设置软件和文件的路径,例如hg38.yml (CIRIquant/hg38.yml,截图如下)。您需要修改内容以适应您的设置(也可以创建另一个yml文件)。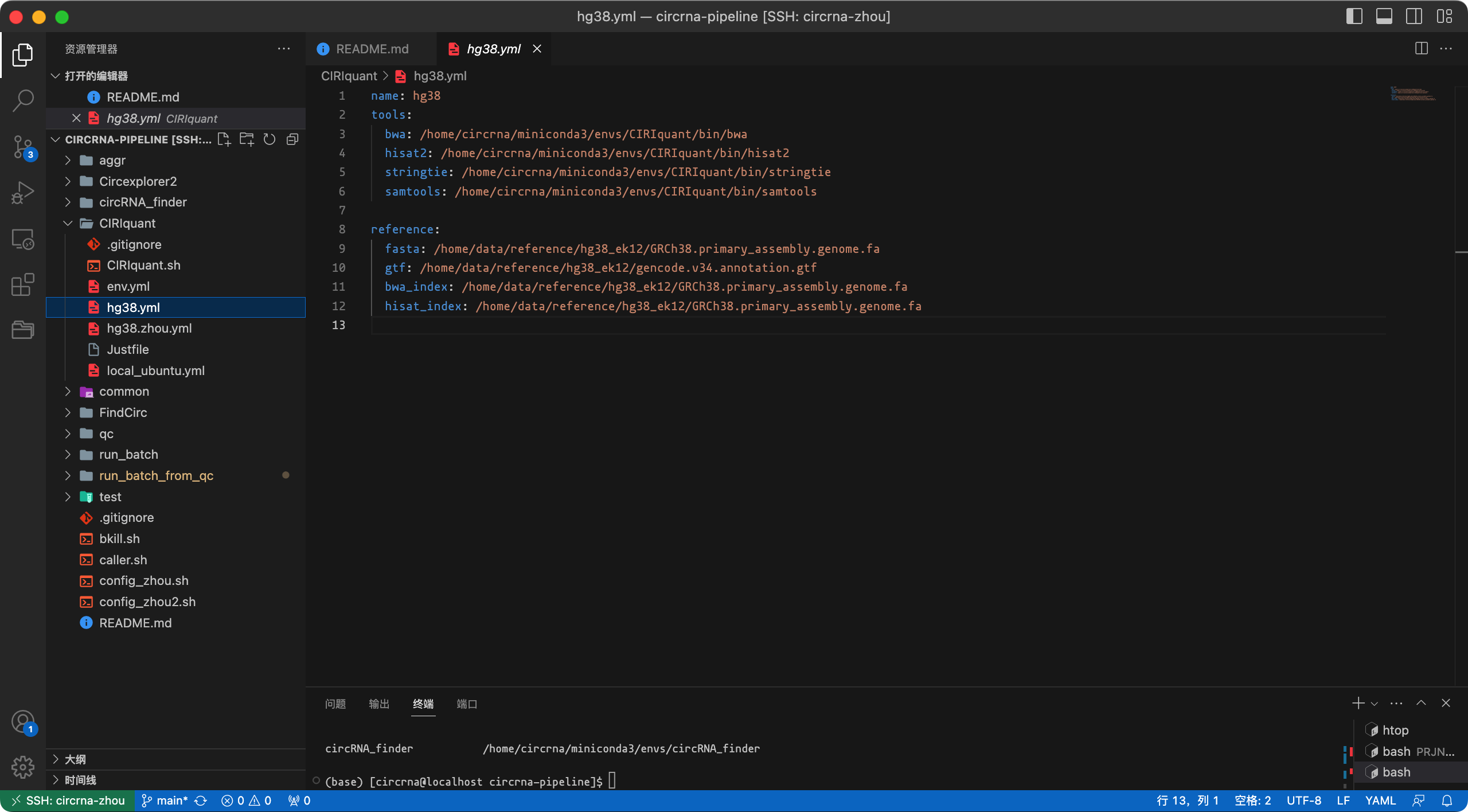
image-20230903160451460 设置一个
config.sh文件,它使用SHELL变量设置了所有必需的设置,config_zhou.sh (config_zhou.sh) 是一个很好的参考(当然,您可以根据需要修改其内容)。image-20230903160258992
使用流程
预处理转录组双端测序的 fastq 文件
这一部分的代码存储在 qc 和common目录下面,我们提供了 md5check.sh 方便用户对自己的数据进行 md5 完整性检查,确保数据完整后可以开始进行数据的预处理步骤,包括质控、去除双端的人为标记序列(adapters)。用户输入的文件后缀需要确保为_1.fastq.gz和_2.fastq.gz。示例文件列表截图如下,红色框选了文件后缀。
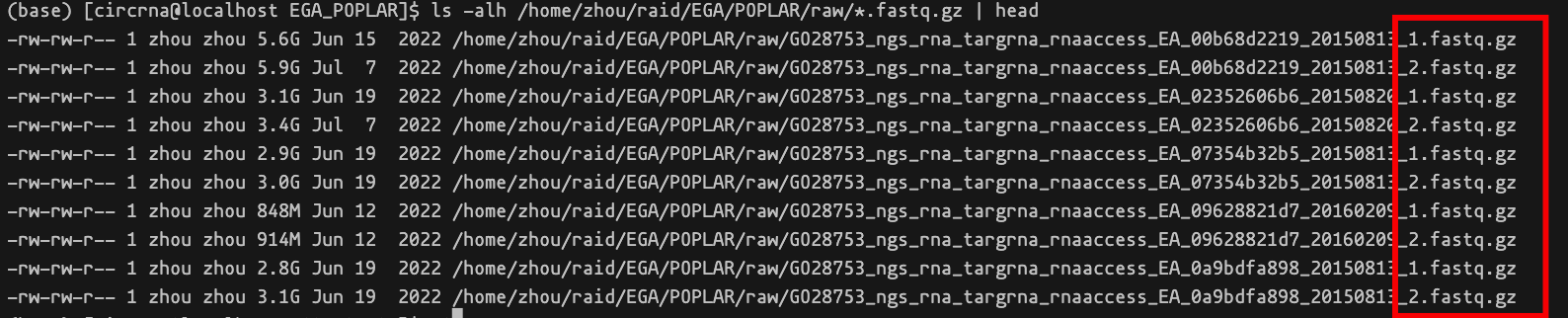
完成后可以进行预处理操作,下面提供了示例的 Shell 脚本。
#!/usr/bin/env bash
# 指定软件路径
py=/home/circrna/miniconda3/bin/python3
fp=/home/circrna/miniconda3/bin/fastp
# 指定代码目录
PIPELINE=/home/circrna/circrna-pipeline
# 指定输入、输出路径
fqfile=./sample_list.txt
indir=/path/to/input
oudir=/path/to/output
# 配置 CPU 数目
nthreads=20
# 调用脚本生成输入目录下的样本 ID 列表
${py} ${PIPELINE}/common/ll_fq.py ${indir} --output ${fqfile}
# 调用预处理命令到后台运行,并将运行的日志导出到 test_qc.log
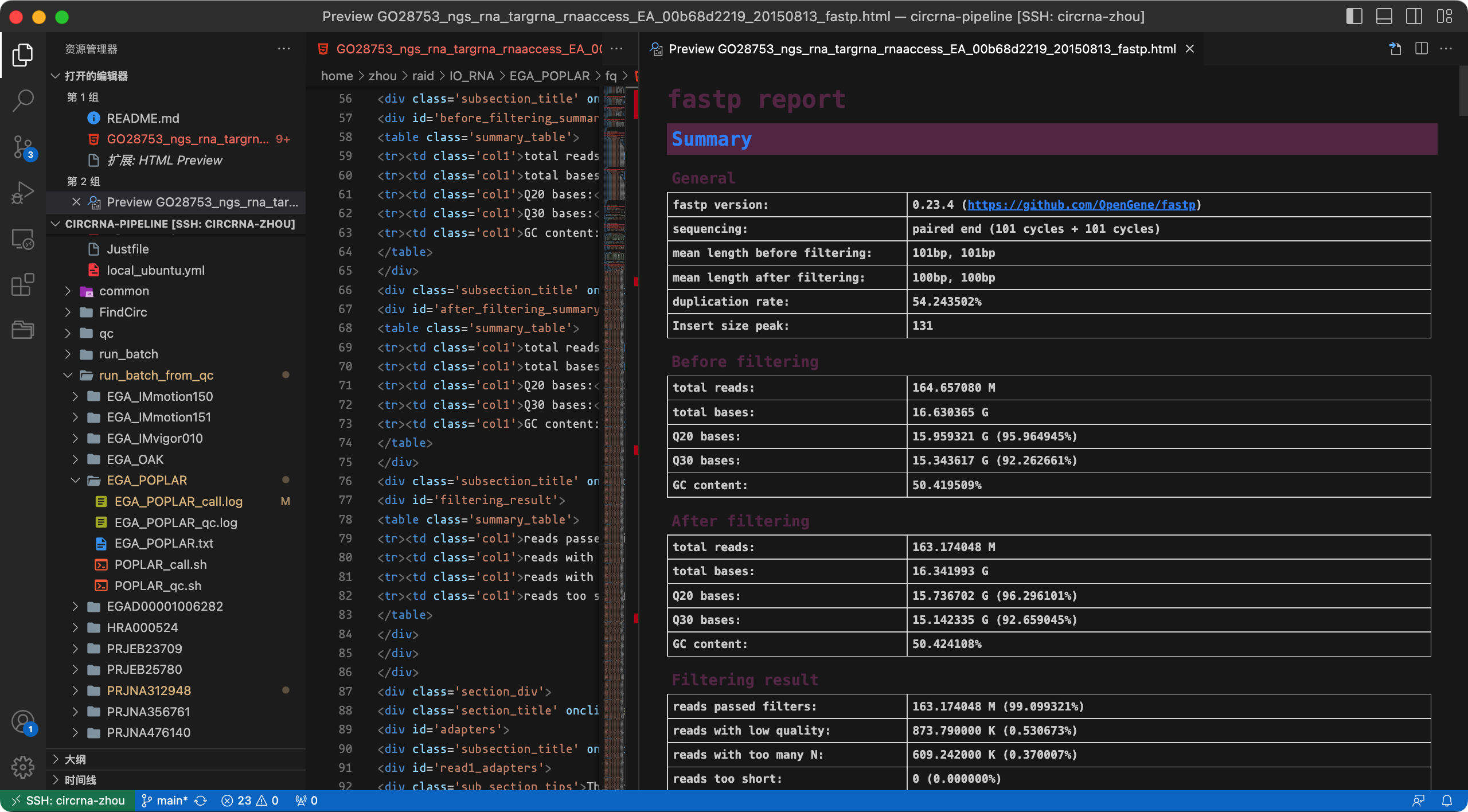
nohup bash ${PIPELINE}/qc/fp.sh ${fqfile} ${indir} ${oudir} ${nthreads} ${fp} &> test_qc.log &下面截图展示了预处理的日志信息,最终会生成样本的质量检测报告(html文件)。
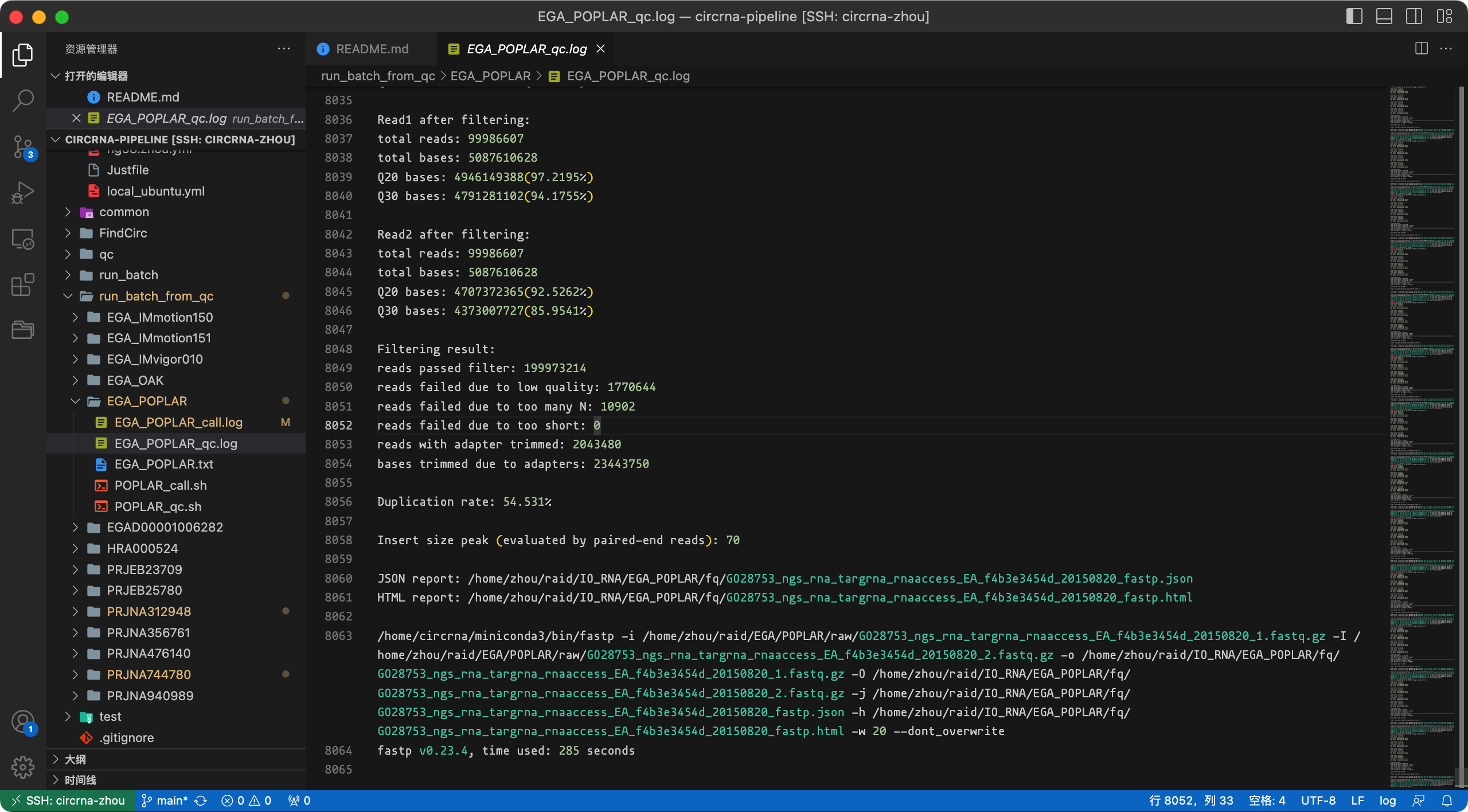
报告文件截图:
运行检测流程
创建一个包含以下设置和命令的shell脚本。
# 指定输入、输出路径
fqfile=./sample_list.txt
indir=/path/include/paired/fastq/files
oudir=/path/to/output
nthreads=20
config=/path/to/your/config.sh
common/ll_fq.py ${indir} --output ${fqfile}
nohup bash caller.sh ${fqfile} ${indir} ${oudir} ${nthreads} ${config} &> run.log &必须在conda
base环境中(或已安装python3的情况下)执行该脚本。 如果您已经自己准备了sample_list.txt文件。 您可以注释掉common/ll_fq.py这一行,然后可以在bash中运行该脚本 而无需任何其他要求(例如,不需要从base环境中安装python3)。
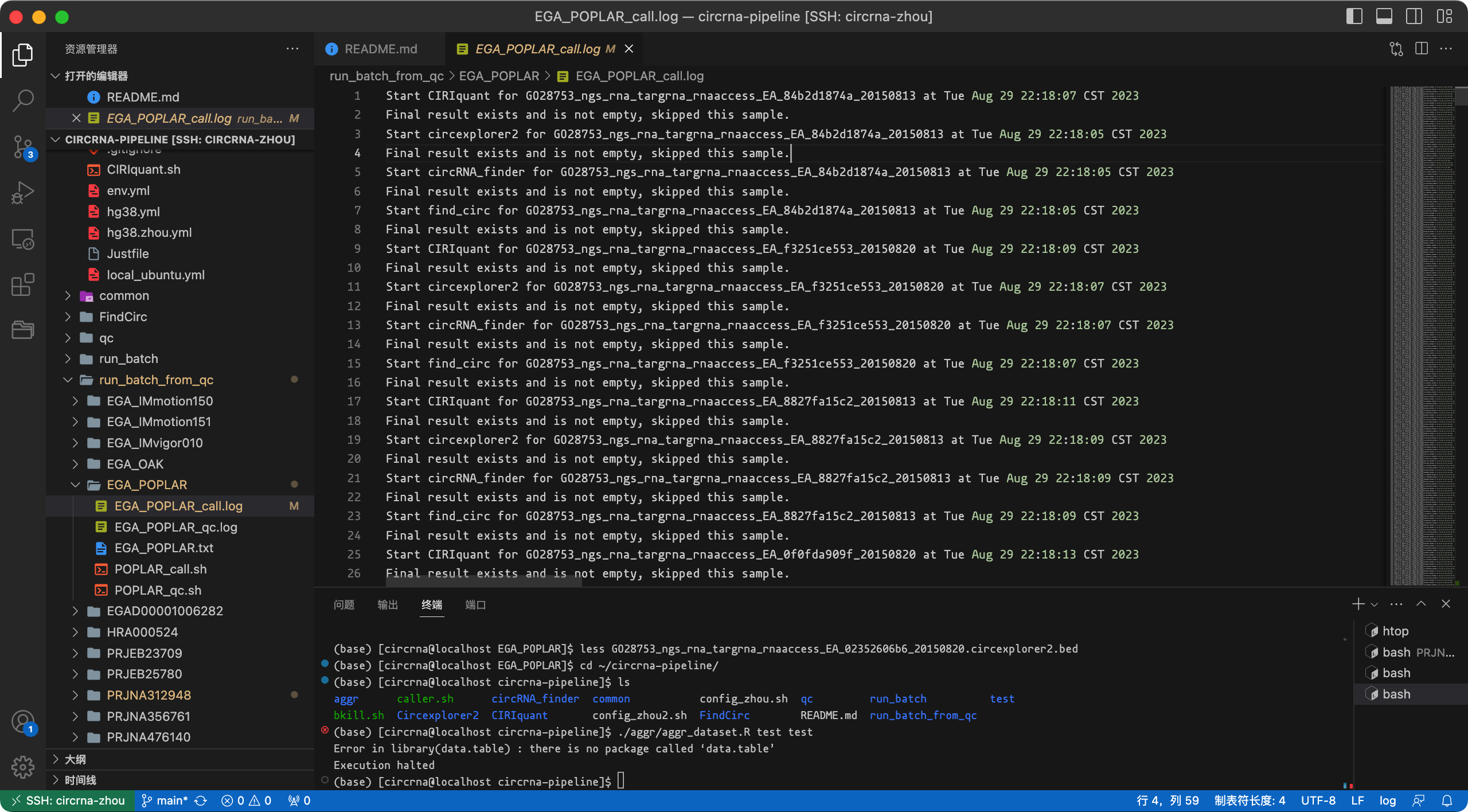
我建议使用2个样本测试流程。如果一切顺利,然后运行您拥有的所有数据文件。流程将跳过已生成结果文件的样本。
日志截图:
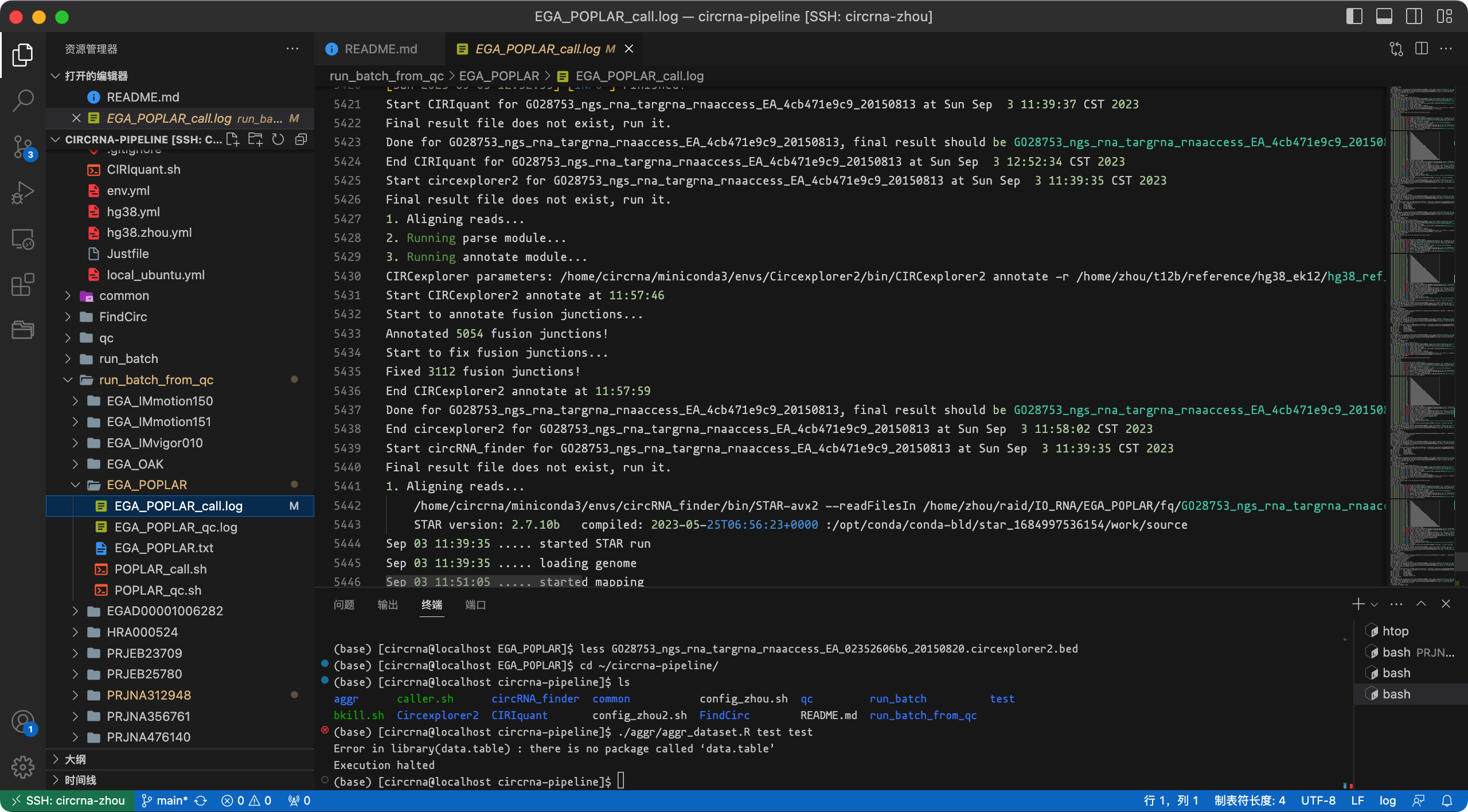
如果运行出现中断,再次运行脚本/命令即可,流程会自动跳过已检测完成的样本。
检查检测输出结果
输出目录包含根据样本名称和方法组合的结果文件。
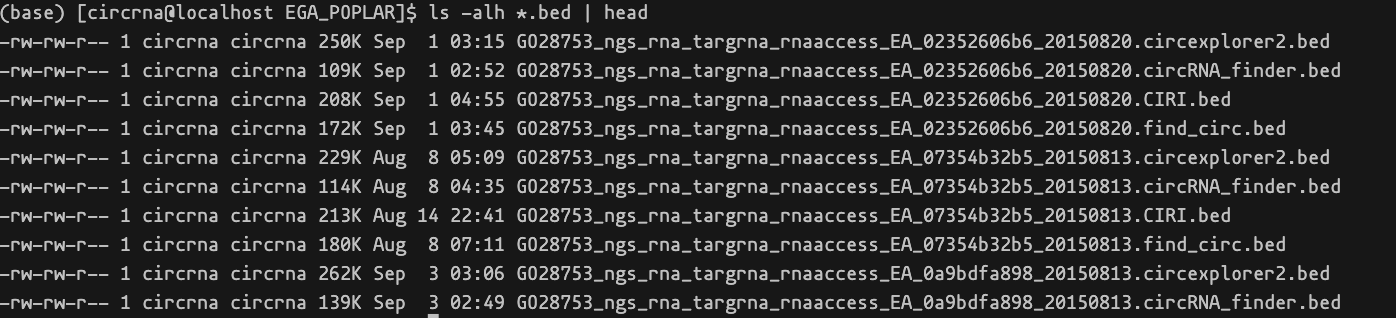
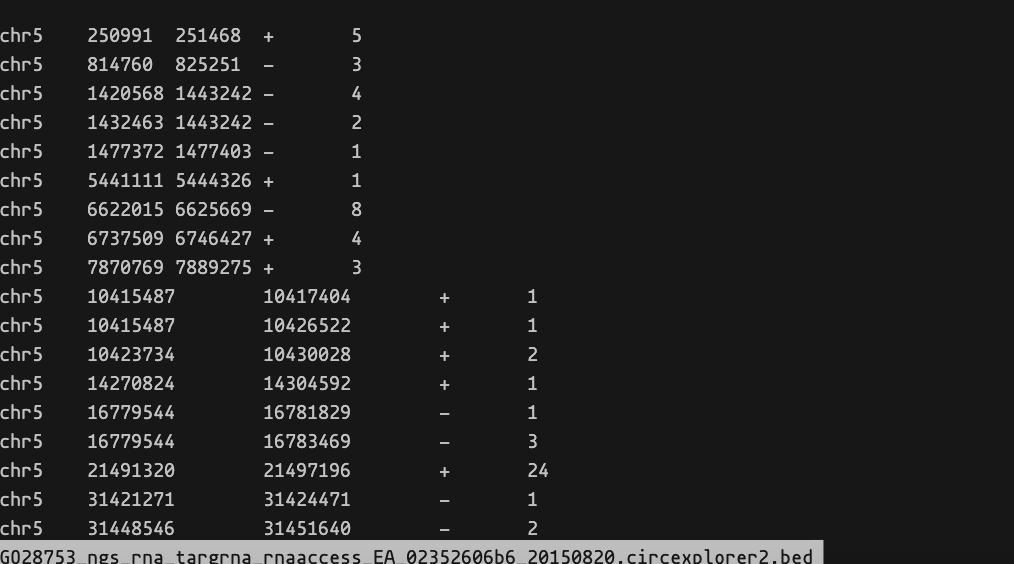
结果文件通常包含循环RNA的位置和计数值。
聚合结果
上述生成了 4 种检测方法的单独结果,我们下一步需要对结果进行聚合,相关代码放在 aggr 目录下。
aggr_beds.R 提供了对方法结果聚合的代码,示例运行命令为:
./aggr/aggr_dataset.R /path/to/result /path/to/aggr_output这样每一个样本都有一个聚合的结果文件。
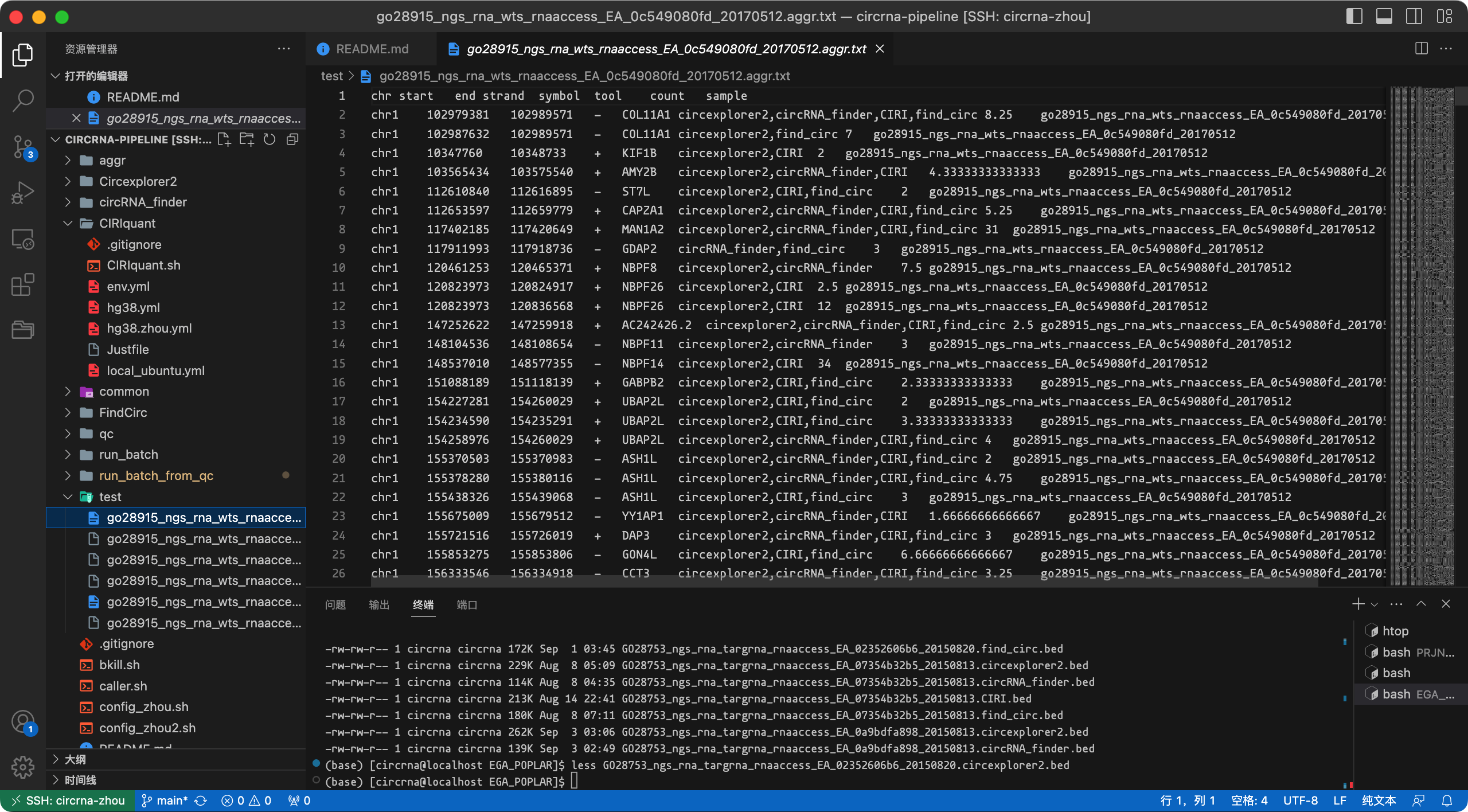
进一步,用户还可以利用 aggr_dataset.R 对全部的样本(即整个输入数据集)结果生成一个整合结果。
调用命令为:
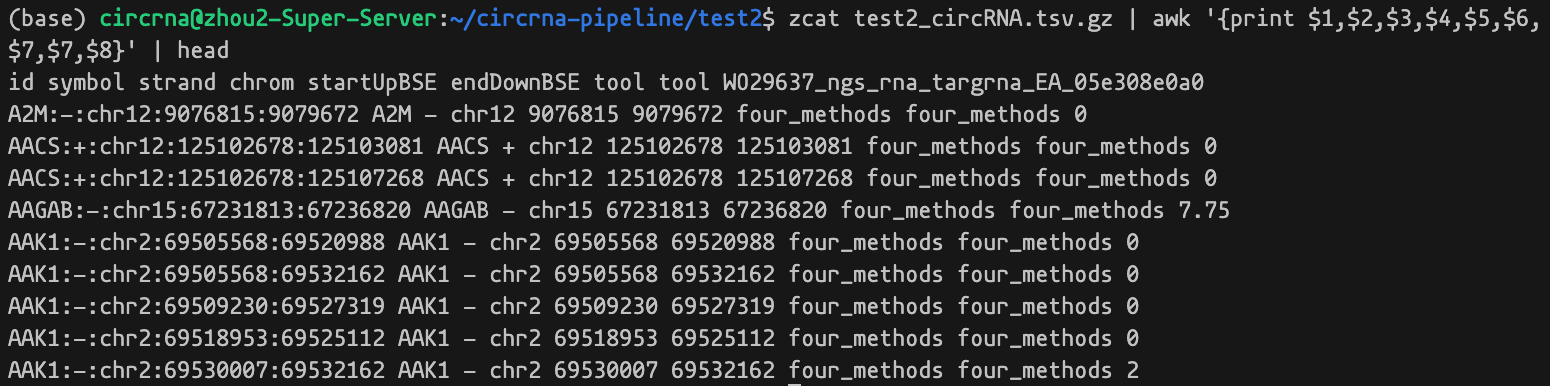
./aggr/aggr_dataset.R /path/to/aggr_output /path/to/dataset_output最后结果会在 /path/to/dataset_output 生成 aggr_output_circRNA.tsv.gz 文件,这是一个大的压缩数据表格,包含以下一些列信息。
id, symbol, strand, chrom, startUpBSE, endDownBSE, tool, samples...- id: circRNA 的唯一标记信息
- symbol: 基因名
- strand: 链信息
- chrom: circRNA 染色体信息
- startUpBSE: circRNA 染色体起点
- endDownBSE: circRNA 染色体终点
- tool: 方法标记
- samples…:其他每列都对应处理的一个样本
下面是测试生成的文件:

查看它的部分内容数据:
用户可以利用流程不同的结果进行下游的分析和研究,包括:
- 不同方法的检测结果
- 同一个样本不同方法检测的聚合结果
- 整个输入样本数据集的检测整合结果