VSHunter: Decode Pattern of Copy Number Profile
Shixiang Wang ShanghaiTech. University
2018-12-06
Source:vignettes/cnPattern.Rmd
cnPattern.RmdThe goal of VSHunter is to capture variation signature from genomic data. For now, we decode copy number pattern from absolute copy number profile. This package collects R code from paper Copy number signatures and mutational processes in ovarian carcinoma and tidy them as a open source R package for bioinformatics community.
Before you use this tool, you have to obtain absolute copy number profile for samples via software like ABSOLUTE v2, QDNASeq etc..
Procedure
- summarise copy-number profile using a number of different feature distributions:
- Sgement size
- Breakpoint number (per ten megabases)
- change-point copy-number
- Breakpoint number (per chromosome arm)
- Length of segments with oscillating copy-number
- apply mixture modelling to breakdown each feature distribution into mixtures of Gaussian or mixtures of Poisson distributions using the flexmix package.
- generate a sample-by-component matrix representing the sum of posterior probabilities of each copy-number event being assigned to each component.
- use NMF package to factorise the sample-by-component matrix into a signature-by-sample matrix and component-by signature-matrix.
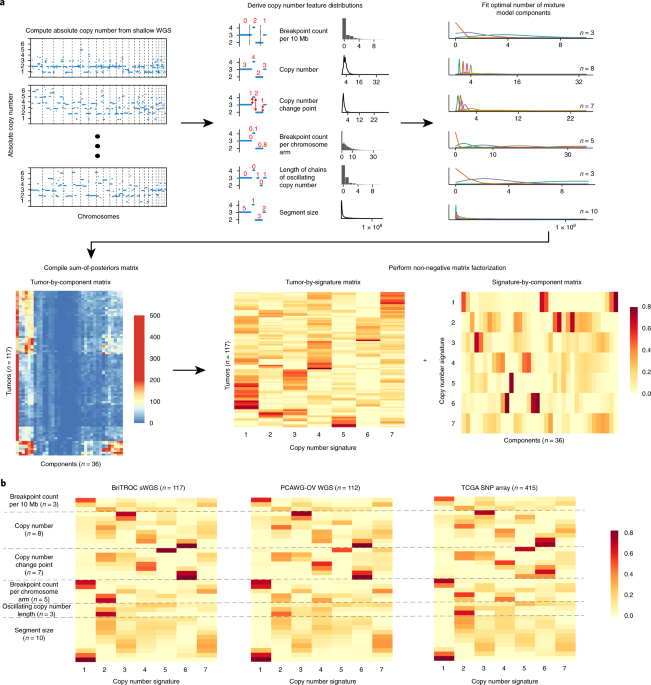
Copy number signature identification, Macintyre, Geoff, et al.(2018)
Installation
You can install UCSCXenaTools from github with:
# install.packages("devtools")
devtools::install_github("ShixiangWang/VSHunter", build_vignettes = TRUE)Load package.
Usage
Load example data:
tcga_segTabs is a list contain absolute copy number profile for multiple samples, each sample is a data.frame in the list.
Generate a sample-by-component matrix
Generate a sample-by-component matrix representing the sum of posterior probabilities of each copy-number event being assigned to each component.
Auto-capture signatures
Function cnv_autoCaptureSignatures() finish three steps (choose best number of signatures, extract signatures and quantify exposure) above in an antomated way. The arguments of this function are same as cnv_chooseSigNumber().
The result object is a list which contains all results need fro downstream analysis, include NMF result related to best rank value, signature matrix, absolute and relative exposure (contribution) and best rank survey etc..
Citation
- Macintyre, Geoff, et al. “Copy number signatures and mutational processes in ovarian carcinoma.” Nature genetics 50.9 (2018): 1262.
If you wanna thank my work for this package, you can also cite (and inlucde link of this package - https://github.com/ShixiangWang/VSHunter):
- Wang, Shixiang, et al. “APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer.” Oncogene (2018).