3 Mutational signature analysis basics
This chapter demonstrates how to run two key mutational signature analyses (de novo signature discovery and signature fitting) in details. More specifically, we will introduce how to identify COSMIC signatures from records of variant calling data. The COSMIC signatures include three type of signatures: SBS, DBS and ID (short for INDEL). For other signature type, please read chapter 4.
The signature identification procedure has been divided into 3 steps:
- Read mutation data.
- Tally components: for SBS, it means classifying SBS records into 96 components (the most common case) and generating sample matrix by counting component in each sample.
- de novo extract signatures or quantify signature activity by fitting observed data to reference signature.
- For de novo signature discovery, there are manual approach and automatic approach. When you choose manual approach, you should estimate signature number and then extract specified number of signatures.
3.1 Data input
The input data should be in VCF, MAF format.
- For VCF, it can only be VCF file paths.
- For MAF, it can be either a MAF file or a
data.frame.
MAF format is the standard way to represent small-scale variants in sigminer. There is a popular R/Bioconductor package maftools (Mayakonda et al. 2018) for analyzing MAF data. It provides an R class MAF to represent MAF format data.
3.1.1 VCF as input
If you use VCF files as input, you can use read_vcf() to read multiple VCF files as a MAF object.
library(sigminer)
vcfs <- list.files(system.file("extdata", package = "sigminer"), "*.vcf", full.names = TRUE)
maf <- read_vcf(vcfs)
## Reading file(s): /Users/wsx/Library/R/sigminer/extdata/test1.vcf, /Users/wsx/Library/R/sigminer/extdata/test2.vcf, /Users/wsx/Library/R/sigminer/extdata/test3.vcf
## It seems /Users/wsx/Library/R/sigminer/extdata/test2.vcf has no normal VCF header, try parsing without header.
## Annotating Variant Type...
## Downloading https://zenodo.org/record/4771552/files/human_hg19_gene_info.rds to /Users/wsx/Library/R/sigminer/extdata/human_hg19_gene_info.rds
## Annotating mutations to first matched gene based on database /Users/wsx/Library/R/sigminer/extdata/human_hg19_gene_info.rds...
## Transforming into a MAF object...
## -Validating
## --Non MAF specific values in Variant_Classification column:
## Unknown
## -Summarizing
## -Processing clinical data
## --Missing clinical data
## -Finished in 0.033s elapsed (0.032s cpu)
maf <- read_vcf(vcfs, keep_only_pass = FALSE)
## Reading file(s): /Users/wsx/Library/R/sigminer/extdata/test1.vcf, /Users/wsx/Library/R/sigminer/extdata/test2.vcf, /Users/wsx/Library/R/sigminer/extdata/test3.vcf
## It seems /Users/wsx/Library/R/sigminer/extdata/test2.vcf has no normal VCF header, try parsing without header.
## Annotating Variant Type...
## Annotating mutations to first matched gene based on database /Users/wsx/Library/R/sigminer/extdata/human_hg19_gene_info.rds...
## Transforming into a MAF object...
## -Validating
## --Non MAF specific values in Variant_Classification column:
## Unknown
## -Summarizing
## -Processing clinical data
## --Missing clinical data
## -Finished in 0.025s elapsed (0.024s cpu)3.1.2 MAF as input
MAF format is the most recommended input, you can provide it either as a file or as a data.frame.
Typically, you can obtain the data in the following ways:
- You get multiple VCF files and convert them into a MAF file (vcf2maf is the most used tool for conversion).
- You get a MAF file from a reference or a public data portal, e.g., cBioPortal or GDC portal.
- You get a EXCEL file providing MAF-like data from a reference, you should read the data firstly (with
readxl::read_excel()) and then construct adata.frameproviding necessary columns.
Once a MAF file or a MAF-like data.frame is ready, you can read/convert it as a MAF object with read_maf(). Here TCGA LAML dataset is used as an example:
laml.maf <- system.file("extdata", "tcga_laml.maf.gz", package = "maftools", mustWork = TRUE)
laml <- read_maf(maf = laml.maf)
## -Reading
## -Validating
## -Silent variants: 475
## -Summarizing
## -Processing clinical data
## --Missing clinical data
## -Finished in 0.295s elapsed (0.271s cpu)
laml
## An object of class MAF
## ID summary Mean Median
## 1: NCBI_Build 37 NA NA
## 2: Center genome.wustl.edu NA NA
## 3: Samples 193 NA NA
## 4: nGenes 1241 NA NA
## 5: Frame_Shift_Del 52 0.269 0
## 6: Frame_Shift_Ins 91 0.472 0
## 7: In_Frame_Del 10 0.052 0
## 8: In_Frame_Ins 42 0.218 0
## 9: Missense_Mutation 1342 6.953 7
## 10: Nonsense_Mutation 103 0.534 0
## 11: Splice_Site 92 0.477 0
## 12: total 1732 8.974 9The laml is a MAF object. The MAF class is exported from maftools to sigminer. So laml can be directly use functions provided by maftools.
As a MAF object, the mutation records are stored in slot data and maf.silent.
head(laml@data)
## Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome
## 1: ABCA10 10349 genome.wustl.edu 37 17
## 2: ABCA4 24 genome.wustl.edu 37 1
## 3: ABCB11 8647 genome.wustl.edu 37 2
## 4: ABCC3 8714 genome.wustl.edu 37 17
## 5: ABCF1 23 genome.wustl.edu 37 6
## 6: ABCG4 64137 genome.wustl.edu 37 11
## Start_Position End_Position Strand Variant_Classification Variant_Type
## 1: 67170917 67170917 + Splice_Site SNP
## 2: 94490594 94490594 + Missense_Mutation SNP
## 3: 169780250 169780250 + Missense_Mutation SNP
## 4: 48760974 48760974 + Missense_Mutation SNP
## 5: 30554429 30554429 + Missense_Mutation SNP
## 6: 119031351 119031351 + Missense_Mutation SNP
## Reference_Allele Tumor_Seq_Allele1 Tumor_Seq_Allele2 Tumor_Sample_Barcode
## 1: T T C TCGA-AB-2988
## 2: C C T TCGA-AB-2869
## 3: G G A TCGA-AB-3009
## 4: C C T TCGA-AB-2887
## 5: G G A TCGA-AB-2920
## 6: A A G TCGA-AB-2934
## Protein_Change i_TumorVAF_WU i_transcript_name
## 1: p.K960R 45.66000 NM_080282.3
## 2: p.R1517H 38.12000 NM_000350.2
## 3: p.A1283V 46.97218 NM_003742.2
## 4: p.P1271S 56.41000 NM_003786.1
## 5: p.G658S 40.95000 NM_001025091.1
## 6: p.Y567C 32.84000 NM_022169.1
head(laml@maf.silent)
## Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome
## 1: ABCC11 85320 genome.wustl.edu 37 16
## 2: ACAN 176 genome.wustl.edu 37 15
## 3: ACAT1 38 genome.wustl.edu 37 11
## 4: ACCN2 41 genome.wustl.edu 37 12
## 5: ACTA2 59 genome.wustl.edu 37 10
## 6: ACTL9 284382 genome.wustl.edu 37 19
## Start_Position End_Position Strand Variant_Classification Variant_Type
## 1: 48244997 48244997 + Silent SNP
## 2: 89401084 89401084 + Silent SNP
## 3: 108009744 108009744 + Silent SNP
## 4: 50452780 50452780 + Silent SNP
## 5: 90695109 90695109 + Silent SNP
## 6: 8808551 8808551 + Silent SNP
## Reference_Allele Tumor_Seq_Allele1 Tumor_Seq_Allele2 Tumor_Sample_Barcode
## 1: G G A TCGA-AB-2830
## 2: C C T TCGA-AB-2898
## 3: T T G TCGA-AB-2887
## 4: C C G TCGA-AB-3009
## 5: C C T TCGA-AB-2973
## 6: G G A TCGA-AB-2936
## Protein_Change i_TumorVAF_WU i_transcript_name
## 1: p.I490I 34.2700000 NM_032583.3
## 2: p.S1756S 38.3000000 NM_013227.2
## 3: p.T185T 49.0400000 NM_000019.3
## 4: p.L77L 48.1000000 NM_020039.2
## 5: p.P335P 0.2012072 NM_001613.1
## 6: p.F167F 46.1500000 NM_178525.3The data slot contains non-silent variants, and the maf.silent slot contains silent variants.
Default uses “Variant Classifications” with high/moderate variant consequences as non-silent variants. http://asia.ensembl.org/Help/Glossary?id=535: “Frame_Shift_Del”, “Frame_Shift_Ins”, “Splice_Site”, “Translation_Start_Site”,“Nonsense_Mutation”, “Nonstop_Mutation”, “In_Frame_Del”,“In_Frame_Ins”, “Missense_Mutation” (see ?read_maf). If you want to change, please set vc_nonSyn option.
Other slots in MAF object are summary data either by sample or gene/variant type etc.
slotNames(laml)
## [1] "data" "variants.per.sample"
## [3] "variant.type.summary" "variant.classification.summary"
## [5] "gene.summary" "summary"
## [7] "maf.silent" "clinical.data"Acute myeloid leukemia is not a good object to study mutational signatures due to low mutation burden, we will use a subset of TCGA breast cohort as for illustration of the following analyses.
Anand Mayakonda has already stored whole TCGA mutation data as MAF objects in TCGAmutations package. Here I will load the TCGA BRCA cohort and create a sub-cohort with 100 tumors.
set.seed(1234)
# brca <- readRDS("data/BRCA.RDs")
brca <- tcga_load("BRCA")
brca <- subsetMaf(brca,
tsb = as.character(sample(brca@variants.per.sample$Tumor_Sample_Barcode, 100))
)
saveRDS(brca, file = "data/brca.rds")Here we save this cohort so no need to download the dataset every time.
brca <- readRDS("data/brca.rds")For CNV and genome rearrangement records, check chapter 4.
3.2 Tally components
3.2.1 The most common 96 components
According to 3-nucleotide context (mutated base, 5’ and 3’ adjacent bases) and base complementary pairing principle, we can divide all SBS mutations into 96 mutation types. We call each mutation type as a component here.
This classification is based the six substitution subtypes: C>A, C>G, C>T, T>A, T>C, and T>G (all substitutions are referred to by the pyrimidine of the mutated Watson—Crick base pair). Further, each of the substitutions is examined by incorporating information on the bases immediately 5’ and 3’ to each mutated base generating 96 possible mutation types (6 types of substitution x 4 types of 5’ base x 4 types of 3’ base).
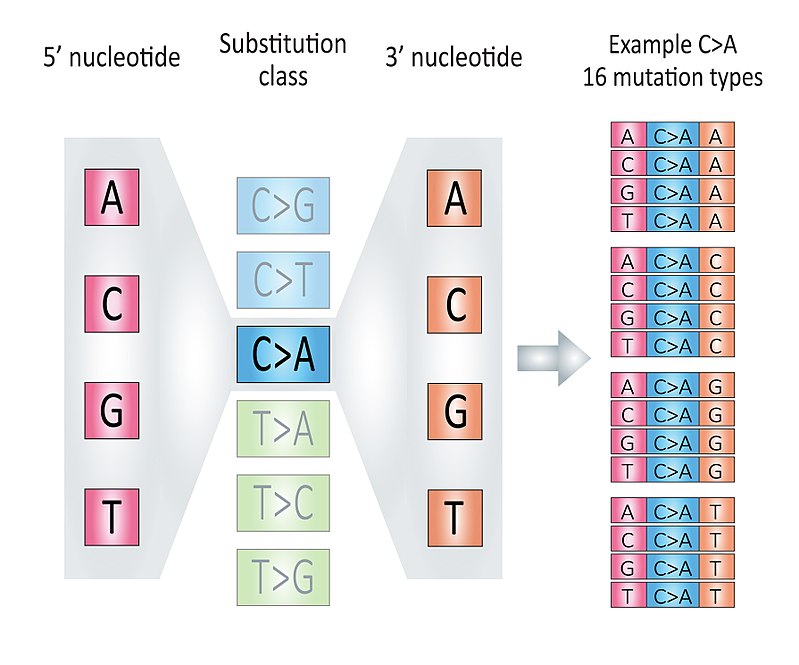
Figure 3.1: The illustration of 96 components, fig source: https://en.wikipedia.org/wiki/Mutational_signatures
We tally components in each sample, and generate a sample-by-component matrix.
mt_tally <- sig_tally(
brca,
ref_genome = "BSgenome.Hsapiens.UCSC.hg19",
use_syn = TRUE
)
## ℹ [2021-12-04 23:49:57]: Started.
## ℹ [2021-12-04 23:50:06]: We would assume you marked all variants' position in + strand.
##
## Attaching package: 'S4Vectors'
## The following objects are masked from 'package:base':
##
## expand.grid, I, unname
##
## Attaching package: 'Biostrings'
## The following object is masked from 'package:base':
##
## strsplit
## ✓ [2021-12-04 23:50:06]: Reference genome loaded.
## ✓ [2021-12-04 23:50:06]: Variants from MAF object queried.
## ✓ [2021-12-04 23:50:06]: Chromosome names checked.
## ✓ [2021-12-04 23:50:06]: Sex chromosomes properly handled.
## ✓ [2021-12-04 23:50:06]: Only variants located in standard chromosomes (1:22, X, Y, M/MT) are kept.
## ✓ [2021-12-04 23:50:06]: Variant start and end position checked.
## ✓ [2021-12-04 23:50:06]: Variant data for matrix generation preprocessed.
## ℹ [2021-12-04 23:50:06]: SBS matrix generation - start.
## ℹ [2021-12-04 23:50:06]: Extracting 5' and 3' adjacent bases.
## ℹ [2021-12-04 23:50:09]: Extracting +/- 20bp around mutated bases for background C>T estimation.
## ℹ [2021-12-04 23:50:09]: Estimating APOBEC enrichment scores.
## ℹ [2021-12-04 23:50:09]: Performing one-way Fisher's test for APOBEC enrichment.
## ✓ [2021-12-04 23:50:10]: APOBEC related mutations are enriched in 28% of samples (APOBEC enrichment score > 2; 28 of 100 samples)
## ℹ [2021-12-04 23:50:10]: Creating SBS sample-by-component matrices.
## ✓ [2021-12-04 23:50:10]: SBS-6 matrix created.
## ✓ [2021-12-04 23:50:10]: SBS-96 matrix created.
## ✓ [2021-12-04 23:50:10]: SBS-1536 matrix created.
## ℹ [2021-12-04 23:50:10]: Return SBS-96 as major matrix.
## ✓ [2021-12-04 23:50:10]: Done.
## ℹ [2021-12-04 23:50:10]: 12.54 secs elapsed.Here set
use_syn = TRUEto include all variant records in MAF object to generate sample matrix.
mt_tally$nmf_matrix[1:5, 1:5]
## A[T>C]A C[T>C]A G[T>C]A T[T>C]A A[C>T]A
## TCGA-A1-A0SH-01A-11D-A099-09 0 0 1 1 0
## TCGA-A2-A04N-01A-11D-A10Y-09 0 0 0 1 2
## TCGA-A2-A0CP-01A-11W-A050-09 0 0 0 0 0
## TCGA-A2-A0EP-01A-52D-A22X-09 0 0 1 0 0
## TCGA-A2-A0EV-01A-11W-A050-09 0 0 1 0 0We use notion left[ref>mut]right to mark each component, e.g. C[T>G]A means a base T with 5’ adjacent base C and 3’ adjacent base A is mutated to base G.
3.2.2 Other Situations
Above we show the most common SBS classifications, there are other situations supported by sigminer, including other classifications for SBS records and other mutation types (DBS and ID). All situations about SBS, DBS and ID signatures are well documented in wiki of SigProfilerMatrixGenerator package.
3.2.2.1 Other SBS classifications
After calling sig_tally(), the most used matrix is stored in nmf_matrix, and all matrices generated by sigminer are stored in all_matrices.
str(mt_tally$all_matrices, max.level = 1)
## List of 3
## $ SBS_6 : int [1:100, 1:6] 7 6 5 4 9 7 5 5 0 5 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_96 : int [1:100, 1:96] 0 0 0 0 0 0 1 2 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_1536: int [1:100, 1:1536] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2If you add the strand classification, all matrices can be generated by sigminer will return.
mt_tally2 <- sig_tally(
brca,
ref_genome = "BSgenome.Hsapiens.UCSC.hg19",
use_syn = TRUE, add_trans_bias = TRUE
)
## ℹ [2021-12-04 23:50:10]: Started.
## ℹ [2021-12-04 23:50:10]: We would assume you marked all variants' position in + strand.
## ✓ [2021-12-04 23:50:10]: Reference genome loaded.
## ✓ [2021-12-04 23:50:10]: Variants from MAF object queried.
## ✓ [2021-12-04 23:50:10]: Chromosome names checked.
## ✓ [2021-12-04 23:50:10]: Sex chromosomes properly handled.
## ✓ [2021-12-04 23:50:10]: Only variants located in standard chromosomes (1:22, X, Y, M/MT) are kept.
## ✓ [2021-12-04 23:50:10]: Variant start and end position checked.
## ✓ [2021-12-04 23:50:10]: Variant data for matrix generation preprocessed.
## ℹ [2021-12-04 23:50:10]: SBS matrix generation - start.
## ℹ [2021-12-04 23:50:10]: Extracting 5' and 3' adjacent bases.
## ℹ [2021-12-04 23:50:11]: Extracting +/- 20bp around mutated bases for background C>T estimation.
## ℹ [2021-12-04 23:50:11]: Estimating APOBEC enrichment scores.
## ℹ [2021-12-04 23:50:11]: Performing one-way Fisher's test for APOBEC enrichment.
## ✓ [2021-12-04 23:50:11]: APOBEC related mutations are enriched in 28% of samples (APOBEC enrichment score > 2; 28 of 100 samples)
## ℹ [2021-12-04 23:50:11]: Creating SBS sample-by-component matrices.
## ✓ [2021-12-04 23:50:11]: SBS-6 matrix created.
## ✓ [2021-12-04 23:50:11]: SBS-96 matrix created.
## ✓ [2021-12-04 23:50:11]: SBS-1536 matrix created.
## ✓ [2021-12-04 23:50:12]: SBS-24 (6x4) matrix created.
## ✓ [2021-12-04 23:50:12]: SBS-384 (96x4) matrix created.
## ✓ [2021-12-04 23:50:12]: SBS-6144 (1536x4) matrix created.
## ℹ [2021-12-04 23:50:12]: Return SBS-192 as major matrix.
## ✓ [2021-12-04 23:50:12]: Done.
## ℹ [2021-12-04 23:50:12]: 2.451 secs elapsed.
str(mt_tally2$all_matrices, max.level = 1)
## List of 7
## $ SBS_6 : int [1:100, 1:6] 7 6 5 4 9 7 5 5 0 5 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_24 : int [1:100, 1:24] 6 3 3 2 6 4 1 2 0 3 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_96 : int [1:100, 1:96] 0 0 0 0 0 0 1 2 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_192 : int [1:100, 1:192] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_384 : int [1:100, 1:384] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_1536: int [1:100, 1:1536] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_6144: int [1:100, 1:6144] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 23.2.2.2 DBS and ID components
If you want to generate DBS or ID matrices, just modify the mode option.
mt_tally_DBS <- sig_tally(
brca,
ref_genome = "BSgenome.Hsapiens.UCSC.hg19",
use_syn = TRUE,
mode = "DBS",
add_trans_bias = TRUE
)
## ℹ [2021-12-04 23:50:13]: Started.
## ℹ [2021-12-04 23:50:13]: We would assume you marked all variants' position in + strand.
## ✓ [2021-12-04 23:50:13]: Reference genome loaded.
## ✓ [2021-12-04 23:50:13]: Variants from MAF object queried.
## ✓ [2021-12-04 23:50:13]: Chromosome names checked.
## ✓ [2021-12-04 23:50:13]: Sex chromosomes properly handled.
## ✓ [2021-12-04 23:50:13]: Only variants located in standard chromosomes (1:22, X, Y, M/MT) are kept.
## ✓ [2021-12-04 23:50:13]: Variant start and end position checked.
## ✓ [2021-12-04 23:50:13]: Variant data for matrix generation preprocessed.
## ℹ [2021-12-04 23:50:13]: DBS matrix generation - start.
## ℹ [2021-12-04 23:50:13]: Searching DBS records...
## ✓ [2021-12-04 23:50:13]: Done.
## ✓ [2021-12-04 23:50:15]: Reference sequences queried from genome.
## ✓ [2021-12-04 23:50:15]: DBS-78 matrix created.
## ✓ [2021-12-04 23:50:15]: DBS-1248 matrix created.
## ✓ [2021-12-04 23:50:15]: DBS-186 matrix created.
## ℹ [2021-12-04 23:50:15]: Return DBS-186 as major matrix.
## ✓ [2021-12-04 23:50:15]: Done.
## ℹ [2021-12-04 23:50:15]: 2.172 secs elapsed.
str(mt_tally_DBS$all_matrices, max.level = 1)
## List of 3
## $ DBS_78 : int [1:100, 1:78] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ DBS_186 : int [1:100, 1:186] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ DBS_1248: int [1:100, 1:1248] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2Program will stop if no records to analyze. Let’s see ID records.
mt_tally_ID <- sig_tally(
brca,
ref_genome = "BSgenome.Hsapiens.UCSC.hg19",
use_syn = TRUE,
mode = "ID",
add_trans_bias = TRUE
)
## ℹ [2021-12-04 23:50:15]: Started.
## ℹ [2021-12-04 23:50:15]: We would assume you marked all variants' position in + strand.
## ✓ [2021-12-04 23:50:15]: Reference genome loaded.
## ✓ [2021-12-04 23:50:15]: Variants from MAF object queried.
## ✓ [2021-12-04 23:50:15]: Chromosome names checked.
## ✓ [2021-12-04 23:50:15]: Sex chromosomes properly handled.
## ✓ [2021-12-04 23:50:15]: Only variants located in standard chromosomes (1:22, X, Y, M/MT) are kept.
## ✓ [2021-12-04 23:50:15]: Variant start and end position checked.
## ✓ [2021-12-04 23:50:15]: Variant data for matrix generation preprocessed.
## ℹ [2021-12-04 23:50:15]: INDEL matrix generation - start.
## ✓ [2021-12-04 23:50:17]: Reference sequences queried from genome.
## ✓ [2021-12-04 23:50:17]: INDEL length extracted.
## ✓ [2021-12-04 23:50:17]: Adjacent copies counted.
## ✓ [2021-12-04 23:50:40]: Microhomology size calculated.
## ✓ [2021-12-04 23:50:40]: INDEL records classified into different components (types).
## ✓ [2021-12-04 23:50:40]: ID-28 matrix created.
## ✓ [2021-12-04 23:50:40]: ID-83 matrix created.
## ✓ [2021-12-04 23:50:40]: ID-415 matrix created.
## ℹ [2021-12-04 23:50:40]: Return ID-415 as major matrix.
## ✓ [2021-12-04 23:50:40]: Done.
## ℹ [2021-12-04 23:50:40]: 24.857 secs elapsed.
str(mt_tally_ID$all_matrices, max.level = 1)
## List of 3
## $ ID_28 : int [1:100, 1:28] 0 1 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ ID_83 : int [1:100, 1:83] 0 1 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ ID_415:'data.frame': 100 obs. of 415 variables:3.2.2.3 Take togother
If you want to get all matrices for SBS, DBS and ID at the same time, you don’t need to write a for loop or type three times to do this.
Just set mode='ALL', sigminer will do it for you!
mt_tally_all <- sig_tally(
brca,
ref_genome = "BSgenome.Hsapiens.UCSC.hg19",
use_syn = TRUE,
mode = "ALL",
add_trans_bias = TRUE
)
## ℹ [2021-12-04 23:50:40]: Started.
## ℹ [2021-12-04 23:50:40]: We would assume you marked all variants' position in + strand.
## ✓ [2021-12-04 23:50:40]: Reference genome loaded.
## ✓ [2021-12-04 23:50:40]: Variants from MAF object queried.
## ✓ [2021-12-04 23:50:40]: Chromosome names checked.
## ✓ [2021-12-04 23:50:40]: Sex chromosomes properly handled.
## ✓ [2021-12-04 23:50:40]: Only variants located in standard chromosomes (1:22, X, Y, M/MT) are kept.
## ✓ [2021-12-04 23:50:40]: Variant start and end position checked.
## ✓ [2021-12-04 23:50:40]: Variant data for matrix generation preprocessed.
## ℹ [2021-12-04 23:50:40]: All types of matrices generation - start.
## ℹ [2021-12-04 23:50:40]: SBS matrix generation - start.
## ℹ [2021-12-04 23:50:40]: Extracting 5' and 3' adjacent bases.
## ℹ [2021-12-04 23:50:42]: Extracting +/- 20bp around mutated bases for background C>T estimation.
## ℹ [2021-12-04 23:50:43]: Estimating APOBEC enrichment scores.
## ℹ [2021-12-04 23:50:43]: Performing one-way Fisher's test for APOBEC enrichment.
## ✓ [2021-12-04 23:50:43]: APOBEC related mutations are enriched in 28% of samples (APOBEC enrichment score > 2; 28 of 100 samples)
## ℹ [2021-12-04 23:50:43]: Creating SBS sample-by-component matrices.
## ✓ [2021-12-04 23:50:43]: SBS-6 matrix created.
## ✓ [2021-12-04 23:50:43]: SBS-96 matrix created.
## ✓ [2021-12-04 23:50:43]: SBS-1536 matrix created.
## ✓ [2021-12-04 23:50:43]: SBS-24 (6x4) matrix created.
## ✓ [2021-12-04 23:50:43]: SBS-384 (96x4) matrix created.
## ✓ [2021-12-04 23:50:43]: SBS-6144 (1536x4) matrix created.
## ℹ [2021-12-04 23:50:43]: Return SBS-192 as major matrix.
## ℹ [2021-12-04 23:50:43]: DBS matrix generation - start.
## ℹ [2021-12-04 23:50:43]: Searching DBS records...
## ✓ [2021-12-04 23:50:44]: Done.
## ✓ [2021-12-04 23:50:45]: Reference sequences queried from genome.
## ✓ [2021-12-04 23:50:45]: DBS-78 matrix created.
## ✓ [2021-12-04 23:50:45]: DBS-1248 matrix created.
## ✓ [2021-12-04 23:50:46]: DBS-186 matrix created.
## ℹ [2021-12-04 23:50:46]: Return DBS-186 as major matrix.
## ℹ [2021-12-04 23:50:46]: INDEL matrix generation - start.
## ✓ [2021-12-04 23:50:48]: Reference sequences queried from genome.
## ✓ [2021-12-04 23:50:48]: INDEL length extracted.
## ✓ [2021-12-04 23:50:48]: Adjacent copies counted.
## ✓ [2021-12-04 23:51:11]: Microhomology size calculated.
## ✓ [2021-12-04 23:51:12]: INDEL records classified into different components (types).
## ✓ [2021-12-04 23:51:12]: ID-28 matrix created.
## ✓ [2021-12-04 23:51:12]: ID-83 matrix created.
## ✓ [2021-12-04 23:51:12]: ID-415 matrix created.
## ℹ [2021-12-04 23:51:12]: Return ID-415 as major matrix.
## ℹ [2021-12-04 23:51:12]: All types of matrices generation (APOBEC scores included) - end.
## ✓ [2021-12-04 23:51:12]: Done.
## ℹ [2021-12-04 23:51:12]: 31.747 secs elapsed.
str(mt_tally_all, max.level = 1)
## List of 14
## $ SBS_6 : int [1:100, 1:6] 7 6 5 4 9 7 5 5 0 5 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_24 : int [1:100, 1:24] 6 3 3 2 6 4 1 2 0 3 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_96 : int [1:100, 1:96] 0 0 0 0 0 0 1 2 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_192 : int [1:100, 1:192] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_384 : int [1:100, 1:384] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_1536 : int [1:100, 1:1536] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ SBS_6144 : int [1:100, 1:6144] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ DBS_78 : int [1:100, 1:78] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ DBS_186 : int [1:100, 1:186] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ DBS_1248 : int [1:100, 1:1248] 0 0 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ ID_28 : int [1:100, 1:28] 0 1 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ ID_83 : int [1:100, 1:83] 0 1 0 0 0 0 0 0 0 0 ...
## ..- attr(*, "dimnames")=List of 2
## $ ID_415 :'data.frame': 100 obs. of 415 variables:
## $ APOBEC_scores:Classes 'data.table' and 'data.frame': 100 obs. of 44 variables:
## ..- attr(*, ".internal.selfref")=<externalptr>
## ..- attr(*, "index")= int(0)
## .. ..- attr(*, "__APOBEC_Enriched")= int [1:100] 17 21 23 24 25 26 27 28 30 32 ...Please note, in this case, just a list containing matrices will return.
3.3 de novo signature discovery
Sigminer provides many approaches to extract mutational signatures. To test their performances, We use 4 mutation catalog datasets (each mutation catalog dataset is composed of 30 samples, 10 COSMIC v2 (SBS) signatures are randomly assigned to each sample with random signature exposure/activity) from Degasperi et al. (2020). The following table shows how many signatures can be recovered and the corresponding average cosine similarity to COSMIC reference signatures for each approach with settings.
| Approach | Selection Way | Setting | Caller | Recommend | Driver | Set1 | Set2 | Set3 | Set4 | Success /Mean | Run time | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Standard NMF | Manual | Default. 50 runs (estimation) + 100 runs (extraction) |
sig_estimate, sig_extract
|
YES ⭐⭐⭐ | R | 10 (0.884) | 10 (0.944) | 9 or 10 (0.998) | 10 (0.994) | ~90%/0.955 | ~1min (8 cores) | This is a basic method, suitable for good mutation data with enough mutations. |
| SigProfiler | Manual/Automatic | Default. 100 runs | sigprofiler_extract |
YES ⭐⭐⭐⭐ | Python/Anaconda | 10 (0.961) | 10 (0.999) | 10 (0.990) | 10 (0.997) | 100%/0.987 | ~1h (8 cores) | A golden standard like approach in this field, but longer run time, and the requirement for Python environment and extra large packages reduce its popularity here. |
| Best Practice | Manual/Automatic | Use bootstrapped catalog (1000 runs) | bp_extract_signatures |
YES ⭐⭐⭐⭐⭐ | R | 10 (0.973) | 10 (0.990) | 10 (0.992) | 10 (0.971) | 100%/0.981 | ~10min (8 cores) | My R implementation for methods from reference #5 and #6. Should be the best option here. (Pay attention to the suggested solution) |
| Best Practice | Manual/Automatic | Use original catalog (1000 runs) | bp_extract_signatures |
NO :star: | R | 10 (0.987) | 9 (0.985) | 10 (0.997) | 9 (0.987) | 50%/0.989 | ~10min (8 cores) | This is created to compare with the approach with bootstrapped catalogs above and the standard NMF way. |
| Bayesian NMF | Automatic | L1KL+optimal (20 runs) | sig_auto_extract |
YES ⭐⭐⭐ | R | 10 (0.994) | 9 (0.997) | 9 (0.998) | 9 (0.999) | 25%/0.997 | ~10min (8 cores) | The Bayesian NMF approach auto reduce the signature number to a proper value from a initial signature number, here is 20. |
| Bayesian NMF | Automatic | L1KL+stable (20 runs) | sig_auto_extract |
YES ⭐⭐⭐⭐ | R | 10 (0.994) | 9 (0.997) | 10 (0.988) | 9 (0.999) | 50%/0.995 | ~10min (8 cores) | See above. |
| Bayesian NMF | Automatic | L2KL+optimal (20 runs) | sig_auto_extract |
NO :star: | R | 12 (0.990) | 13 (0.988) | 12 (0.902) | 12 (0.994) | 0%/0.969 | ~10min (8 cores) | See above. |
| Bayesian NMF | Automatic | L2KL+stable (20 runs) | sig_auto_extract |
NO :star: | R | 12 (0.990) | 12 (0.988) | 12 (0.902) | 12 (0.994) | 0%/0.969 | ~10min (8 cores) | See above. |
| Bayesian NMF | Automatic | L1WL2H+optimal (20 runs) | sig_auto_extract |
YES ⭐⭐⭐ | R | 9 (0.989) | 9 (0.999) | 9 (0.996) | 9 (1.000) | 0%/0.996 | ~10min (8 cores) | See above. |
| Bayesian NMF | Automatic | L1WL2H+stable (20 runs) | sig_auto_extract |
YES ⭐⭐⭐⭐ | R | 9 (0.989) | 9 (0.999) | 9 (0.996) | 9 (1.000) | 0%/0.996 | ~10min (8 cores) | See above. |
NOTE: although Bayesian NMF approach with L1KL or L1WL2H prior cannot recover all 10 signatures here, but it is close to the true answer from initial signature number 20 in a automatic way, and the result signatures are highly similar to reference signatures. This also reminds us that we could not use this method to find signatures with small contributions in tumors.
From sigminer v2.1.0, an unified interface sig_unify_extract(). has been implemented to access the
4 signature extraction approaches.
args(sig_unify_extract)
## function (nmf_matrix, range = 2:5, nrun = 10, approach = c("bayes_nmf",
## "repeated_nmf", "bootstrap_nmf", "sigprofiler"), cores = 1L,
## ...)
## NULLOnce you determine a method, please read all parameters shown in the detail function.
- “bayes_nmf” corresponds to
sig_auto_extract(). - “repeated_nmf” corresponds to
sig_extract(). - “bootstrap_nmf” coresponds to
bp_extract_signatures(). - “sigprofiler” corresponds to
sigprofiler_extract().
Note, when you use sig_extract() (“repeated_nmf”) and you don’t know how
to select the signature number, you should run sig_estimate() firstly.
3.3.1 Manual signature estimation and extraction
For example, let’s try signature number 2-6. For simplicity, we just run NMF 10 times for each signature number. We use 4 cores to speed up the computation.
mt_est <- sig_estimate(mt_tally$nmf_matrix,
range = 2:6,
nrun = 10, # increase this value if you wana a more stable estimation
use_random = FALSE, # if TRUE, add results from randomized input
cores = 4,
verbose = TRUE
)We can show signature number survey for different measures by show_sig_number_survey2().
## You can also select the measures to show
## by 'what' option
show_sig_number_survey2(mt_est$survey)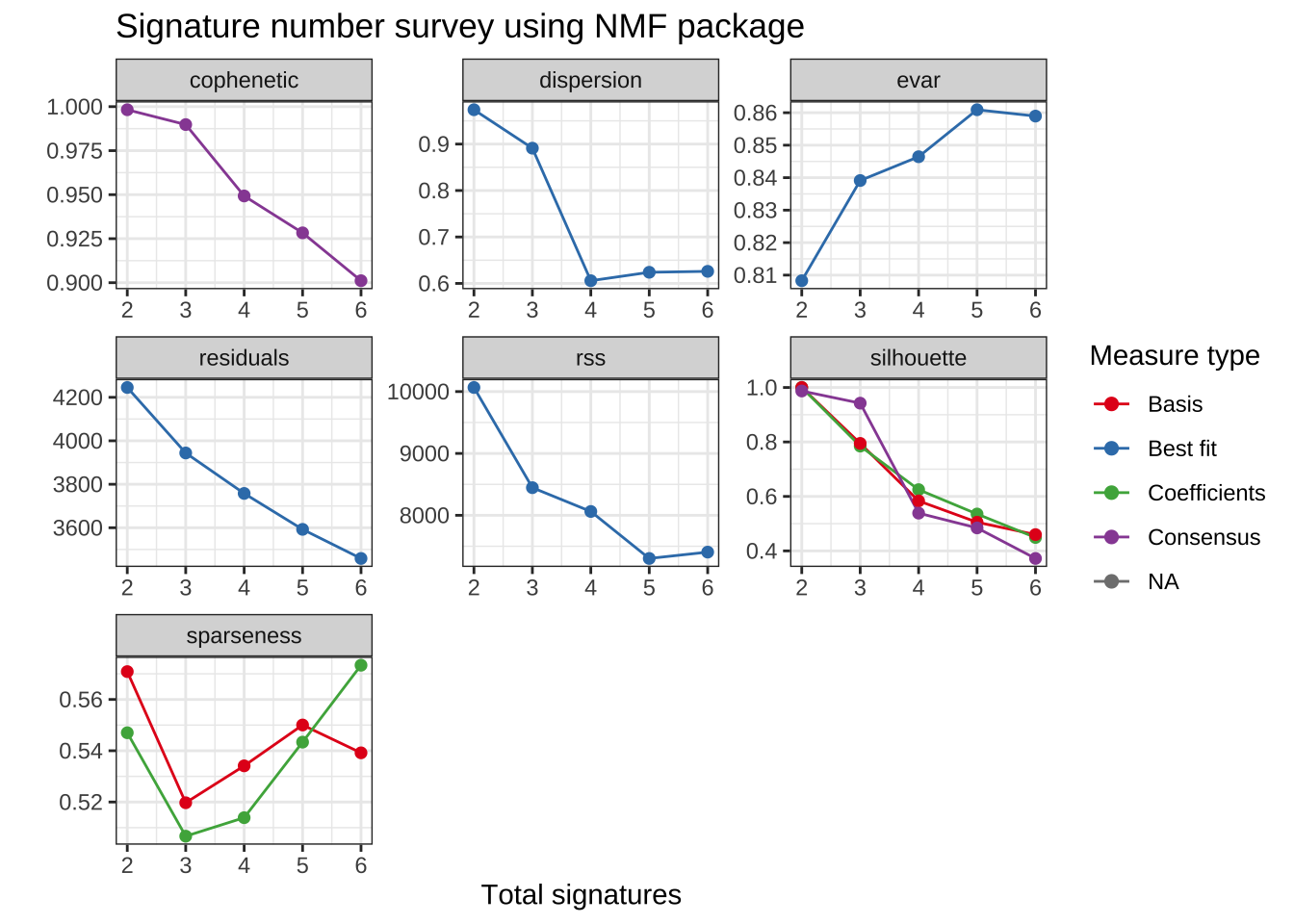
For the details of all the measures above, please read Gaujoux and Seoighe (2010) and vignette of R package NMF. The measures either provide stability (
cophenetic) or how well can be reconstructed (rss).
Typically, measure cophenetic is used for determining the signature number. We can easily generate an elbow plot
with function show_sig_number_survey().
show_sig_number_survey(mt_est$survey, right_y = NULL)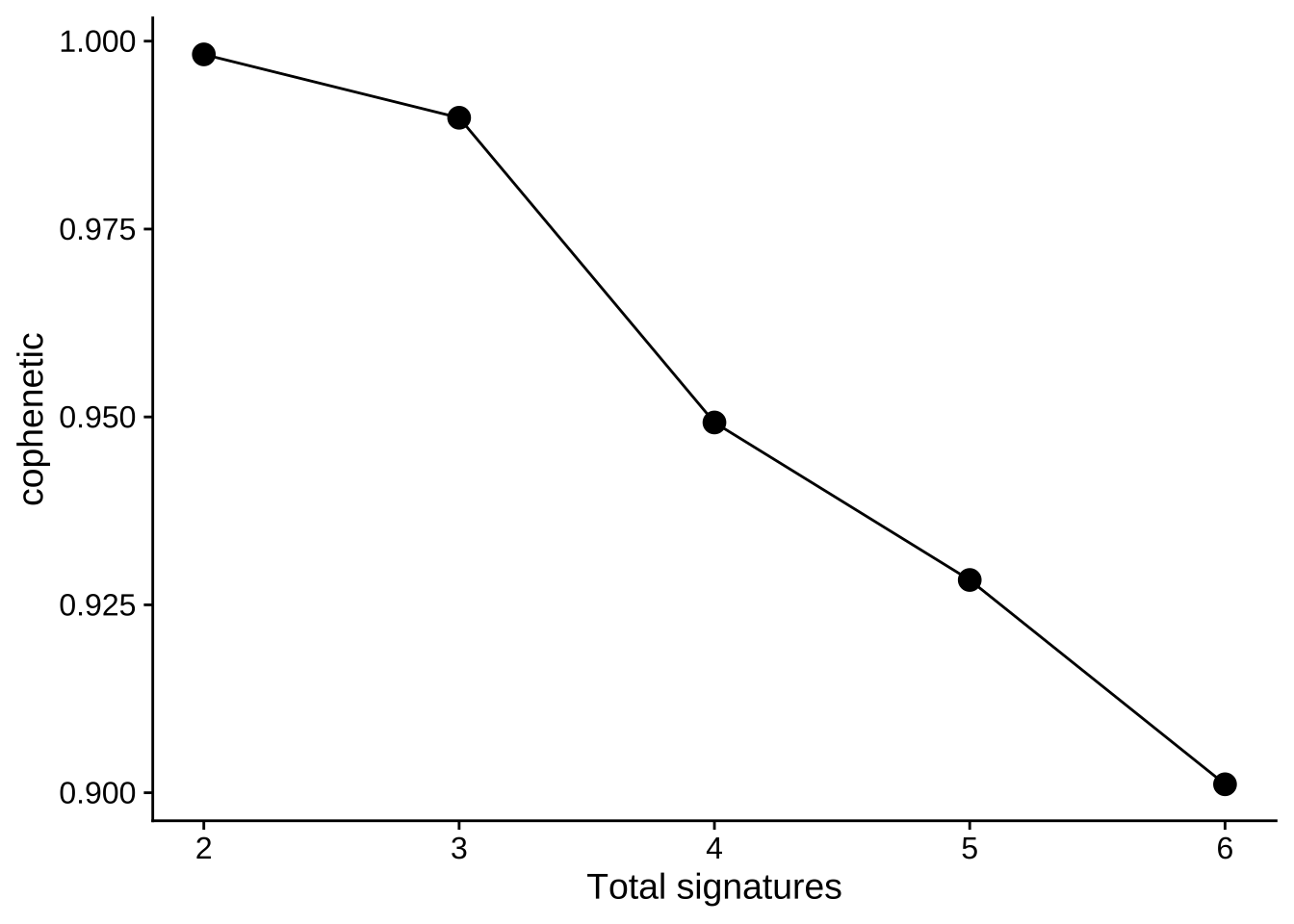
The most common approach is to use the cophenetic correlation coefficient. Brunet et al. suggested choosing the smallest value of r for which this coefficient starts decreasing. (Gaujoux and Seoighe 2010) Cophenetic value (range from 0-1) indicates the robustness of consensus matrix clustering. In this situation, 3 is good. However, we can found that the cophenetic values are all >=0.9 from 2 to 5. So the more suitable way is considering both stability and reconstruction error at the same time, it can be easily done by
show_sig_number_survey().
show_sig_number_survey(mt_est$survey)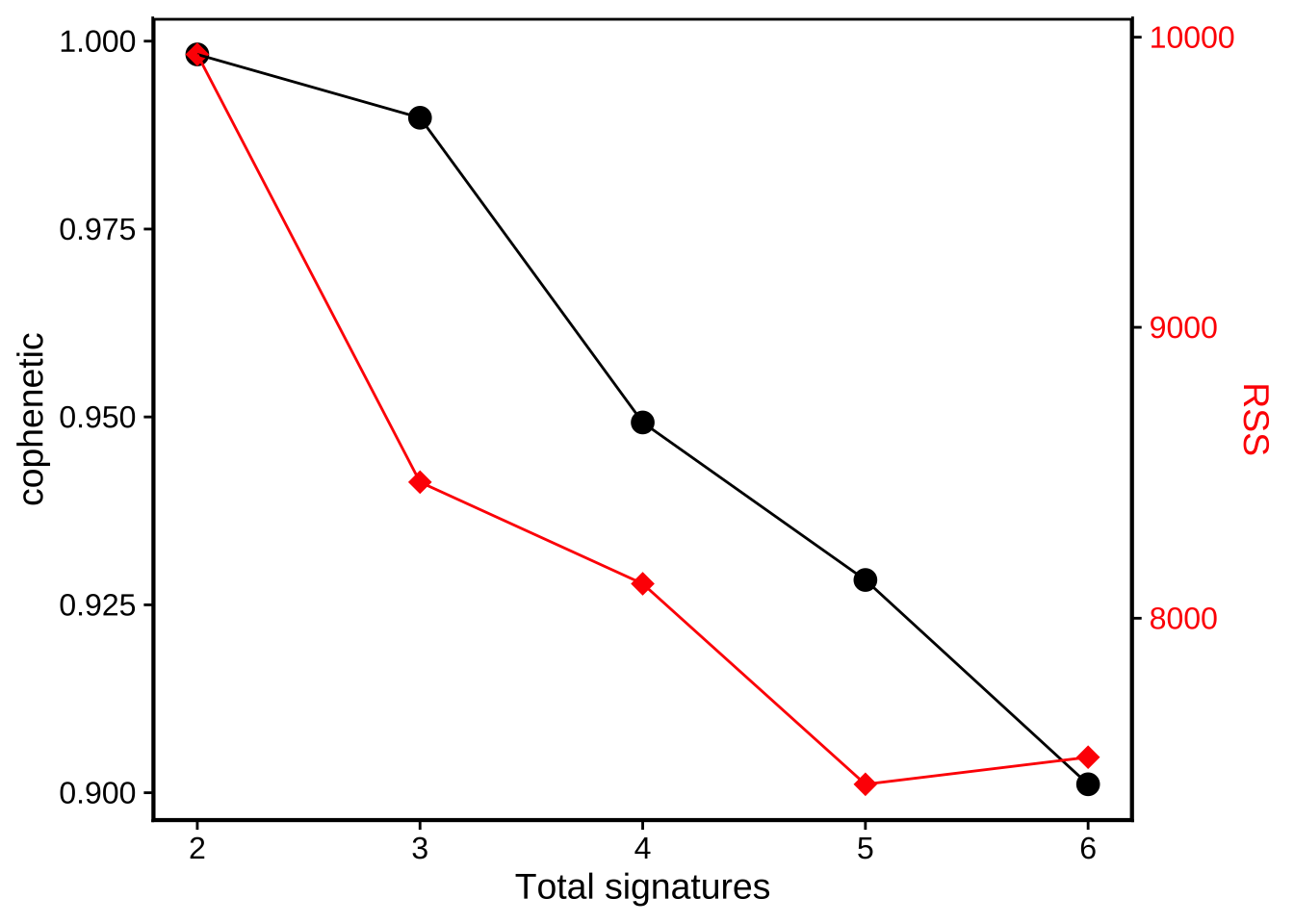
This function is very flexible, you can pick up any measure to the left/right axis. However, the default setting is the most recommended way. We can see that we get a minimal RSS in signature number, and when this value goes from 5 to 6, the RSS increase! So we should not choose signature number more than 5 here because 6 is overfitting.
NOTE: There are no gold standard to determine the signature number. Sometimes, you should consider multiple measures. Remember, the most important thing is that you should have a good biological explanation for each signature. The best solution in study may not be the best solution in math.
Now that the 5 signatures should be a stable solution, next we can extract it with more runs to obtain the optimal result. In general, use 30~50 NMF runs will get a robust result.
mt_sig <- sig_extract(mt_tally$nmf_matrix,
n_sig = 5,
nrun = 30,
cores = 4
)3.3.2 Automatic extraction
If you have no idea to select an optimal signature number from procedures above, you can try auto-extraction approaches provided by sigminer.
The latest version of sigminer provides three ways to auto-extract mutational signatures.
- Auto-extract signatures by automatic relevance determination technique in non-negative matrix factorization (Tan and Févotte 2012), the code is implemented by SignatureAnalyzer (Kim et al. 2016) and exported to sigminer. This approach is known as bayesian NMF and the default approach in
sig_unify_extract(). - Auto-extract signatures by SigProfiler, the gold-standard tool used for identifying signatures cataloged in COSMIC database. The technical details please read Alexandrov et al. (2020).
- Multiple NMF runs with bootstrapped mutation catalogs. This method is adopted from Degasperi et al. (2020).
3.3.2.1 Method 1: Bayesian NMF
In this approach, you need to set a maximum signature number (default is 25) and run times to get the result. 10 for nrun here is okay, and more than 100 is not recommended.
The Bayesian NMF will starts from a larger signature number and reduce it to a proper signature number to maximize posterior probability.
mt_sig2 <- sig_unify_extract(mt_tally$nmf_matrix, range = 10, nrun = 10)This is same as:
mt_sig2 <- sig_auto_extract(mt_tally$nmf_matrix,
K0 = 10, nrun = 10,
strategy = "stable"
)Here the program uses ‘robust’ strategy to return the result (see strategy option). It means that if you run 10 times and 6 of them return 4 signatures, then the optimal result with 4 signatures will be returned.
The info of each run can be given as:
knitr::kable(mt_sig2$Raw$summary_run)| Run | K | posterior | file |
|---|---|---|---|
| 5 | 3 | -1497.498 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.5.rds |
| 3 | 3 | -1497.752 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.3.rds |
| 7 | 3 | -1497.841 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.7.rds |
| 1 | 3 | -1498.525 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.1.rds |
| 9 | 3 | -1498.528 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.9.rds |
| 10 | 3 | -1498.883 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.10.rds |
| 6 | 3 | -1499.748 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.6.rds |
| 4 | 3 | -1499.814 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.4.rds |
| 8 | 3 | -1500.195 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.8.rds |
| 2 | 4 | -1604.000 | /var/folders/qx/5tqxhrrd5xd_n9rfbb_m5c200000gn/T//RtmpPz6VVL/BayesNMF.2.rds |
The mt_sig2 has similar structure as mut_sig.
3.3.2.2 Method 2: SigProfiler
Sigminer provides two functions sigprofiler_extract() and sigprofiler_import() to install, use SigProfiler and import SigProfilerExtractor results into R as a Signature object like other extraction methods mentioned above.
An (not running) example is given below (see ?sigprofiler for more info).
reticulate::conda_list()
sigprofiler_extract(cn_tally_W$nmf_matrix, "~/test/test_sigminer",
use_conda = TRUE
)
# Same as
# sig_unify_extract(mt_tally$nmf_matrix, use_conda = FALSE, py_path = "/Users/wsx/anaconda3/bin/python", approach = "sigprofiler", out = "~/test/test_sigminer")
sigprofiler_extract(mt_tally$nmf_matrix, "~/test/test_sigminer",
use_conda = FALSE, py_path = "/Users/wsx/anaconda3/bin/python"
)3.3.2.3 Method 3: bootstrapped NMF
# Same as
# mt_sig3 <- sig_unify_extract(
# cn_tally_W$nmf_matrix,
# range = 3:8,
# nrun = 10
# n_bootstrap = 5
# )
mt_sig3 <- bp_extract_signatures(
cn_tally_W$nmf_matrix,
range = 3:8,
n_bootstrap = 5,
n_nmf_run = 10
)3.4 Match Signatures
After extracting signatures, we need to know their etiologies. This can be done by comparing the identified signatures and reference signatures from COSMIC database.
sim <- get_sig_similarity(mt_sig2)
## -Comparing against COSMIC signatures
## ------------------------------------
## --Found Sig1 most similar to COSMIC_3
## Aetiology: defects in DNA-DSB repair by HR [similarity: 0.826]
## --Found Sig2 most similar to COSMIC_1
## Aetiology: spontaneous deamination of 5-methylcytosine [similarity: 0.944]
## --Found Sig3 most similar to COSMIC_2
## Aetiology: APOBEC Cytidine Deaminase (C>T) [similarity: 0.838]
## ------------------------------------
## Return result invisiblely.The result object sim is a list.
str(sim)
## List of 4
## $ similarity : num [1:3, 1:30] 0.826 0.274 0.373 0.56 0.944 0.164 0.088 0.259 0.838 0.722 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:3] "Sig1" "Sig2" "Sig3"
## .. ..$ : chr [1:30] "COSMIC_3" "COSMIC_1" "COSMIC_2" "COSMIC_4" ...
## $ aetiology_db:List of 1
## ..$ : chr [1:30] "spontaneous deamination of 5-methylcytosine" "APOBEC Cytidine Deaminase (C>T)" "defects in DNA-DSB repair by HR" "exposure to tobacco (smoking) mutagens" ...
## $ best_match :List of 3
## ..$ Sig1:List of 2
## .. ..$ aetiology : chr "defects in DNA-DSB repair by HR"
## .. ..$ best_match: chr "Best match: COSMIC_3 [similarity: 0.826]"
## ..$ Sig2:List of 2
## .. ..$ aetiology : chr "spontaneous deamination of 5-methylcytosine"
## .. ..$ best_match: chr "Best match: COSMIC_1 [similarity: 0.944]"
## ..$ Sig3:List of 2
## .. ..$ aetiology : chr "APOBEC Cytidine Deaminase (C>T)"
## .. ..$ best_match: chr "Best match: COSMIC_2 [similarity: 0.838]"
## $ rss : num [1:3, 1:30] 0.04294 0.00684 0.14741 0.24881 0.24076 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:3] "Sig1" "Sig2" "Sig3"
## .. ..$ : chr [1:30] "COSMIC_1" "COSMIC_2" "COSMIC_3" "COSMIC_4" ...
## - attr(*, "class")= chr [1:2] "similarity" "list"From the result we can see that three signatures are properly matched to COSMIC reference signatures. If you find unknown signatures in your study, you should explore the etiologies by other analyses and even experiments.
The similarity matrix can be plotted.
pheatmap::pheatmap(sim$similarity)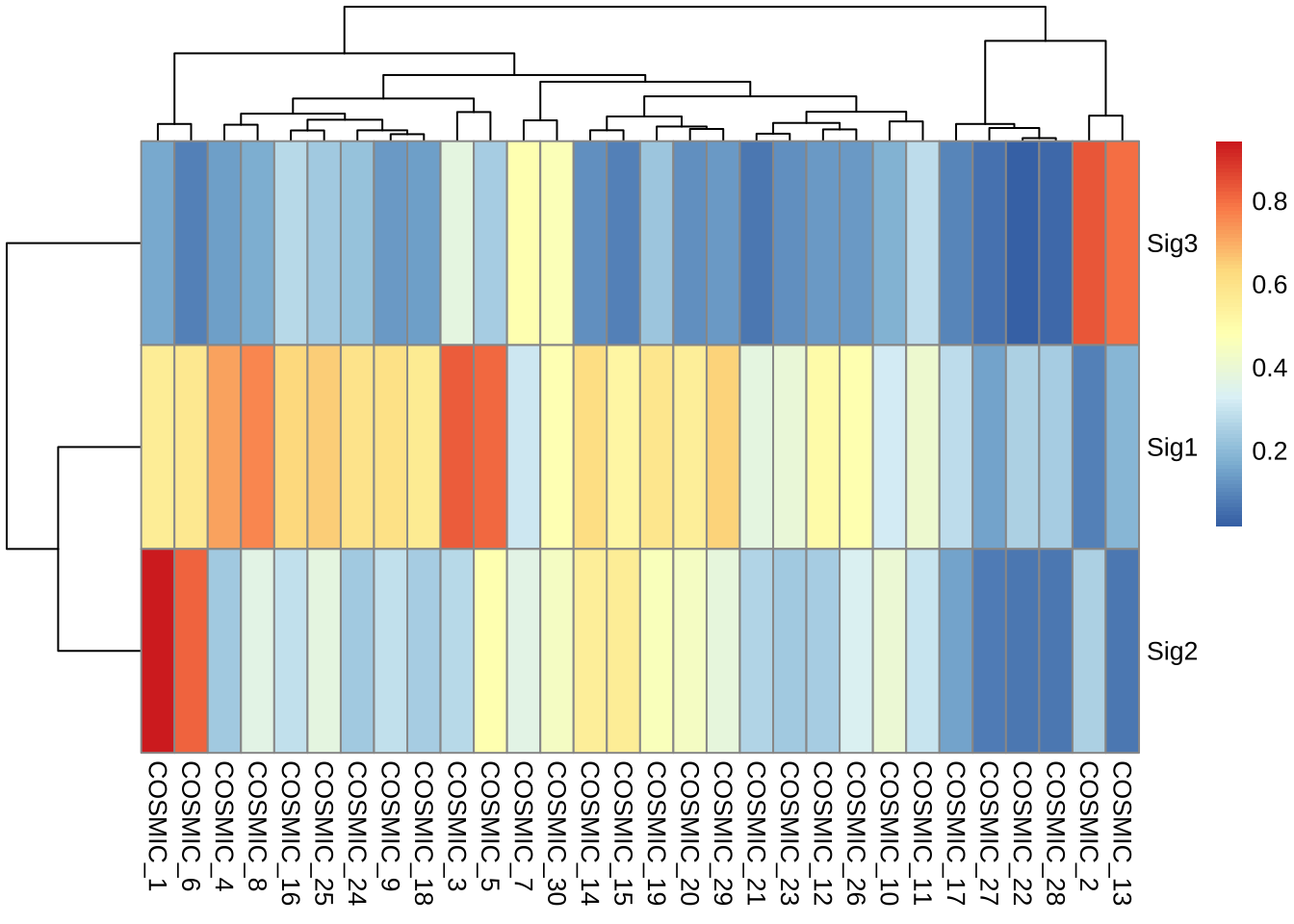
You can also try the COSMIC signature database V3 with:
sim_v3 <- get_sig_similarity(mt_sig2, sig_db = "SBS")
## -Comparing against COSMIC signatures
## ------------------------------------
## --Found Sig1 most similar to SBS3
## Aetiology: Defective homologous recombination DNA damage repair [similarity: 0.828]
## --Found Sig2 most similar to SBS1
## Aetiology: Spontaneous deamination of 5-methylcytosine (clock-like signature) [similarity: 0.876]
## --Found Sig3 most similar to SBS2
## Aetiology: Activity of APOBEC family of cytidine deaminases [similarity: 0.746]
## ------------------------------------
## Return result invisiblely.3.5 Reference signature fitting
Besides de novo signature discovery shown in previous chapters, another common task is that
you have gotten some reference signatures (either from known database like COSMIC or de novo discovery step), you want to know how these signatures contribute (fit) in a sample. That’s the target of sig_fit().
sig_fit() uses multiple methods to compute exposure of pre-defined signatures from the spectrum of a (can be more) sample. Use ?sig_fit see more detail.
To show how this function works, we use a sample with maximum mutation counts as example data.
i <- which.max(apply(mt_tally$nmf_matrix, 1, sum))
example_mat <- mt_tally$nmf_matrix[i, , drop = FALSE] %>% t()
head(example_mat)
## TCGA-A8-A09G-01A-21W-A019-09
## A[T>C]A 1
## C[T>C]A 0
## G[T>C]A 1
## T[T>C]A 1
## A[C>T]A 5
## C[C>T]A 33.5.1 Fit signatures from reference signature databasase
For SBS signatures, users may want to directly use reference signatures from COSMIC database.
sig_fit(example_mat, sig_index = 1:30)
## ℹ [2021-12-04 23:51:15]: Started.
## ✓ [2021-12-04 23:51:15]: Signature index detected.
## ℹ [2021-12-04 23:51:15]: Checking signature database in package.
## ℹ [2021-12-04 23:51:15]: Checking signature index.
## ℹ [2021-12-04 23:51:15]: Valid index for db 'legacy':
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
## ✓ [2021-12-04 23:51:15]: Database and index checked.
## ✓ [2021-12-04 23:51:15]: Signature normalized.
## ℹ [2021-12-04 23:51:15]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:15]: Checked.
## ℹ [2021-12-04 23:51:15]: Checking rownames for catalog matrix and signature matrix.
## ℹ [2021-12-04 23:51:15]: Matrix V and W don't have same orders. Try reordering...
## ✓ [2021-12-04 23:51:15]: Checked.
## ✓ [2021-12-04 23:51:15]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:15]: Corresponding function generated.
## ℹ [2021-12-04 23:51:15]: Calling function.
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A8-A09G-01A-21W-A019-09
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: 0.046 secs elapsed.
## TCGA-A8-A09G-01A-21W-A019-09
## COSMIC_1 24.215933
## COSMIC_2 127.164108
## COSMIC_3 0.000000
## COSMIC_4 0.000000
## COSMIC_5 0.000000
## COSMIC_6 0.000000
## COSMIC_7 4.907674
## COSMIC_8 0.000000
## COSMIC_9 0.000000
## COSMIC_10 3.584276
## COSMIC_11 0.000000
## COSMIC_12 11.062526
## COSMIC_13 168.298139
## COSMIC_14 0.000000
## COSMIC_15 0.000000
## COSMIC_16 0.000000
## COSMIC_17 5.578495
## COSMIC_18 0.000000
## COSMIC_19 0.000000
## COSMIC_20 0.000000
## COSMIC_21 0.000000
## COSMIC_22 0.000000
## COSMIC_23 0.000000
## COSMIC_24 12.084656
## COSMIC_25 0.000000
## COSMIC_26 0.000000
## COSMIC_27 0.000000
## COSMIC_28 0.000000
## COSMIC_29 0.000000
## COSMIC_30 0.104192At default, COSMIC v2 signature database with 30 reference signatures is used (i.e.
sig_db = "legacy"). Setsig_db = "SBS"for COSMIC v3 signature database. That’s it!
You can set type = "relative" for getting relative exposure.
sig_fit(example_mat, sig_index = 1:30, type = "relative")
## ℹ [2021-12-04 23:51:15]: Started.
## ✓ [2021-12-04 23:51:15]: Signature index detected.
## ℹ [2021-12-04 23:51:15]: Checking signature database in package.
## ℹ [2021-12-04 23:51:15]: Checking signature index.
## ℹ [2021-12-04 23:51:15]: Valid index for db 'legacy':
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
## ✓ [2021-12-04 23:51:15]: Database and index checked.
## ✓ [2021-12-04 23:51:15]: Signature normalized.
## ℹ [2021-12-04 23:51:15]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:15]: Checked.
## ℹ [2021-12-04 23:51:15]: Checking rownames for catalog matrix and signature matrix.
## ℹ [2021-12-04 23:51:15]: Matrix V and W don't have same orders. Try reordering...
## ✓ [2021-12-04 23:51:15]: Checked.
## ✓ [2021-12-04 23:51:15]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:15]: Corresponding function generated.
## ℹ [2021-12-04 23:51:15]: Calling function.
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A8-A09G-01A-21W-A019-09
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: 0.034 secs elapsed.
## TCGA-A8-A09G-01A-21W-A019-09
## COSMIC_1 0.067832
## COSMIC_2 0.356202
## COSMIC_3 0.000000
## COSMIC_4 0.000000
## COSMIC_5 0.000000
## COSMIC_6 0.000000
## COSMIC_7 0.013747
## COSMIC_8 0.000000
## COSMIC_9 0.000000
## COSMIC_10 0.010040
## COSMIC_11 0.000000
## COSMIC_12 0.030987
## COSMIC_13 0.471423
## COSMIC_14 0.000000
## COSMIC_15 0.000000
## COSMIC_16 0.000000
## COSMIC_17 0.015626
## COSMIC_18 0.000000
## COSMIC_19 0.000000
## COSMIC_20 0.000000
## COSMIC_21 0.000000
## COSMIC_22 0.000000
## COSMIC_23 0.000000
## COSMIC_24 0.033851
## COSMIC_25 0.000000
## COSMIC_26 0.000000
## COSMIC_27 0.000000
## COSMIC_28 0.000000
## COSMIC_29 0.000000
## COSMIC_30 0.000292For multiple samples, you can return a data.table, it can be easier to integrate with other information in R.
sig_fit(t(mt_tally$nmf_matrix[1:5, ]), sig_index = 1:30, return_class = "data.table", rel_threshold = 0.05)
## ℹ [2021-12-04 23:51:15]: Started.
## ✓ [2021-12-04 23:51:15]: Signature index detected.
## ℹ [2021-12-04 23:51:15]: Checking signature database in package.
## ℹ [2021-12-04 23:51:15]: Checking signature index.
## ℹ [2021-12-04 23:51:15]: Valid index for db 'legacy':
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
## ✓ [2021-12-04 23:51:15]: Database and index checked.
## ✓ [2021-12-04 23:51:15]: Signature normalized.
## ℹ [2021-12-04 23:51:15]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:15]: Checked.
## ℹ [2021-12-04 23:51:15]: Checking rownames for catalog matrix and signature matrix.
## ℹ [2021-12-04 23:51:15]: Matrix V and W don't have same orders. Try reordering...
## ✓ [2021-12-04 23:51:15]: Checked.
## ✓ [2021-12-04 23:51:15]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:15]: Corresponding function generated.
## ℹ [2021-12-04 23:51:15]: Calling function.
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A1-A0SH-01A-11D-A099-09
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A2-A04N-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A2-A0CP-01A-11W-A050-09
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A2-A0EP-01A-52D-A22X-09
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A2-A0EV-01A-11W-A050-09
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: 0.054 secs elapsed.
## sample COSMIC_1 COSMIC_2 COSMIC_3 COSMIC_4 COSMIC_5
## 1: TCGA-A1-A0SH-01A-11D-A099-09 0.000000 37.420603 13.78689 0.000000 0
## 2: TCGA-A2-A04N-01A-11D-A10Y-09 20.039543 2.888675 0.00000 0.000000 0
## 3: TCGA-A2-A0CP-01A-11W-A050-09 3.648658 0.000000 0.00000 7.083113 0
## 4: TCGA-A2-A0EP-01A-52D-A22X-09 0.000000 0.000000 0.00000 2.492218 0
## 5: TCGA-A2-A0EV-01A-11W-A050-09 6.458422 0.000000 14.83102 0.000000 0
## COSMIC_6 COSMIC_7 COSMIC_8 COSMIC_9 COSMIC_10 COSMIC_11 COSMIC_12 COSMIC_13
## 1: 12.93472 21.332013 0.00000 0 0.000000 0 0.000000 31.306430
## 2: 0.00000 6.865345 12.11501 0 0.000000 0 0.000000 0.000000
## 3: 0.00000 10.348536 0.00000 0 0.000000 0 0.000000 0.000000
## 4: 0.00000 2.156319 0.00000 0 0.000000 0 1.334731 4.654227
## 5: 14.78142 21.963952 0.00000 0 7.978962 0 0.000000 5.713563
## COSMIC_14 COSMIC_15 COSMIC_16 COSMIC_17 COSMIC_18 COSMIC_19 COSMIC_20
## 1: 0.000000 0.00000 0 0 12.007682 0 0.000000
## 2: 0.000000 0.00000 0 0 0.000000 0 7.516444
## 3: 0.000000 18.37734 0 0 4.384106 0 0.000000
## 4: 6.728415 0.00000 0 0 0.000000 0 0.000000
## 5: 0.000000 0.00000 0 0 0.000000 0 0.000000
## COSMIC_21 COSMIC_22 COSMIC_23 COSMIC_24 COSMIC_25 COSMIC_26 COSMIC_27
## 1: 0.000000 0 0.00000 0 0 0 0
## 2: 0.000000 0 0.00000 0 0 0 0
## 3: 0.000000 0 0.00000 0 0 0 0
## 4: 0.000000 0 1.26778 0 0 0 0
## 5: 4.311951 0 0.00000 0 0 0 0
## COSMIC_28 COSMIC_29 COSMIC_30
## 1: 0 0.000000 0
## 2: 0 0.000000 0
## 3: 0 4.776321 0
## 4: 0 0.000000 0
## 5: 0 0.000000 0When you set multiple signatures, we recommend setting rel_threshold option, which will set exposure of a signature to 0 if its relative exposure in a sample less than the rel_threshold.
3.5.2 Fit custom signatures
We have already determined the SBS signatures before. Here we can set them to sig option.
sig_fit(example_mat, sig = mt_sig2)
## ℹ [2021-12-04 23:51:15]: Started.
## ℹ [2021-12-04 23:51:15]: Signature index not detected.
## ✓ [2021-12-04 23:51:15]: Signature object detected.
## ✓ [2021-12-04 23:51:15]: Database and index checked.
## ✓ [2021-12-04 23:51:15]: Signature normalized.
## ℹ [2021-12-04 23:51:15]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:15]: Checked.
## ℹ [2021-12-04 23:51:15]: Checking rownames for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:15]: Checked.
## ✓ [2021-12-04 23:51:15]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:15]: Corresponding function generated.
## ℹ [2021-12-04 23:51:15]: Calling function.
## ℹ [2021-12-04 23:51:15]: Fitting sample: TCGA-A8-A09G-01A-21W-A019-09
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:15]: Done.
## ℹ [2021-12-04 23:51:15]: 0.03 secs elapsed.
## TCGA-A8-A09G-01A-21W-A019-09
## Sig1 0
## Sig2 0
## Sig3 3573.5.3 Performance comparison
Now that we can use sig_fit for getting optimal exposures, we can compare the RSS between raw matrix and the reconstructed matrix either by NMF and sig_fit().
i.e.
\[ RSS = \sum(\hat H - H)^2 \]
## Exposure got from NMF
sum((apply(mt_sig2$Signature, 2, function(x) x / sum(x)) %*% mt_sig2$Exposure - t(mt_tally$nmf_matrix))^2)
## [1] 8890.449
## Exposure optimized by sig_fit
H_estimate <- apply(mt_sig2$Signature, 2, function(x) x / sum(x)) %*% sig_fit(t(mt_tally$nmf_matrix), sig = mt_sig2)
## ℹ [2021-12-04 23:51:16]: Started.
## ℹ [2021-12-04 23:51:16]: Signature index not detected.
## ✓ [2021-12-04 23:51:16]: Signature object detected.
## ✓ [2021-12-04 23:51:16]: Database and index checked.
## ✓ [2021-12-04 23:51:16]: Signature normalized.
## ℹ [2021-12-04 23:51:16]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:16]: Checked.
## ℹ [2021-12-04 23:51:16]: Checking rownames for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:16]: Checked.
## ✓ [2021-12-04 23:51:16]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:16]: Corresponding function generated.
## ℹ [2021-12-04 23:51:16]: Calling function.
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A1-A0SH-01A-11D-A099-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A04N-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0CP-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0EP-01A-52D-A22X-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0EV-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0SX-01A-12D-A099-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0T7-01A-21D-A099-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A0YF-01A-21D-A10G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A25F-01A-11D-A167-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A3XW-01A-11D-A23C-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A2-A4S1-01A-21D-A25Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A7-A0D9-01A-31W-A071-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A7-A13F-01A-11D-A12Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A7-A5ZV-01A-11D-A28B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A06P-01A-11W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A076-01A-21W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A07W-01A-11W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A084-01A-21W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A08S-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A09G-01A-21W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A0A4-01A-11W-A019-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A0AB-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AC-A2B8-01A-11D-A17D-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AC-A2FO-01A-11D-A17W-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AC-A3YI-01A-21D-A23C-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AC-A8OS-01A-12D-A41F-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AN-A0FK-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AN-A0FT-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AN-A0XO-01A-11D-A10G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AO-A1KS-01A-11D-A13L-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AQ-A54O-01A-11D-A25Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AQ-A7U7-01A-22D-A351-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A0TP-01A-11D-A099-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A0U3-01A-11D-A10G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A1AH-01A-11D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A1AJ-01A-21D-A12Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A1AN-01A-11D-A12Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A24N-01A-11D-A167-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A252-01A-11D-A167-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A2LL-01A-11D-A17W-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-AR-A2LO-01A-31D-A18P-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0IE-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0IM-01A-11W-A050-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0IP-01A-11D-A045-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0RV-01A-11D-A099-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0WZ-01A-11D-A10G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A0X1-01A-11D-A10G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A1KC-01B-11D-A159-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A401-01A-11D-A23C-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-B6-A40C-01A-11D-A23C-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0AV-01A-31D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0BT-01A-11D-A12Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0DL-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0DO-01B-11D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0DT-01A-21D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0GY-01A-11W-A071-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A0H6-01A-21W-A071-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A18K-01A-11D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A1FU-01A-11D-A14G-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A202-01A-11D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A5IZ-01A-11D-A27P-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A6R8-01A-21D-A33E-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-BH-A8G0-01A-11D-A351-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-C8-A131-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A147-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1JG-01B-11D-A13L-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1JH-01A-11D-A188-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1JJ-01A-31D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1JT-01A-31D-A13L-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1JU-01A-11D-A13L-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1X7-01A-11D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1X8-01A-11D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A1XL-01A-11D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-D8-A27V-01A-12D-A17D-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A108-01A-13D-A10M-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A10F-01A-11D-A10M-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A14T-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A152-01A-11D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A15D-01A-11D-A10Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A15L-01A-11D-A12B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A1BD-01A-11D-A12Q-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A1IH-01A-11D-A188-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A1II-01A-11D-A142-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A1IJ-01A-11D-A142-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A1L6-01A-11D-A13L-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E2-A9RU-01A-11D-A41F-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E9-A1NE-01A-21D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E9-A22A-01A-11D-A159-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E9-A22E-01A-11D-A159-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E9-A3QA-01A-61D-A228-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-E9-A5FL-01A-11D-A27P-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-EW-A1PA-01A-11D-A142-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-EW-A1PH-01A-11D-A14K-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-GM-A2DB-01A-31D-A19Y-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-LD-A9QF-01A-32D-A41F-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-LL-A5YP-01A-21D-A28B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-LL-A73Z-01A-11D-A32I-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-OL-A5RY-01A-21D-A28B-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-PE-A5DD-01A-12D-A27P-09
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-S3-AA17-01A-11D-A41F-09
## ✓ [2021-12-04 23:51:16]: Done.
## ℹ [2021-12-04 23:51:16]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:16]: Done.
## ℹ [2021-12-04 23:51:16]: 0.19 secs elapsed.
H_estimate <- apply(H_estimate, 2, function(x) ifelse(is.nan(x), 0, x))
H_real <- t(mt_tally$nmf_matrix)
sum((H_estimate - H_real)^2)
## [1] 8237.8323.5.4 Estimate exposure/activity stability by bootstrapping
This feature is based on sig_fit(), it uses the resampling data of original input and runs sig_fit() multiple times to estimate the exposure. Bootstrapped replicates >= 100 is recommended, here I just use 10 times for illustration.
bt_result <- sig_fit_bootstrap_batch(example_mat, sig = mt_sig2, n = 10)
## ℹ [2021-12-04 23:51:16]: Batch Bootstrap Signature Exposure Analysis Started.
## ℹ [2021-12-04 23:51:16]: Samples to be filtered out:
## ℹ [2021-12-04 23:51:16]: Finding optimal exposures (&errors) for different methods.
## ℹ [2021-12-04 23:51:16]: Calling method `QP`.
## ℹ [2021-12-04 23:51:16]: Started.
## ℹ [2021-12-04 23:51:16]: Signature index not detected.
## ✓ [2021-12-04 23:51:16]: Signature object detected.
## ✓ [2021-12-04 23:51:16]: Database and index checked.
## ✓ [2021-12-04 23:51:16]: Signature normalized.
## ℹ [2021-12-04 23:51:16]: Checking row number for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:16]: Checked.
## ℹ [2021-12-04 23:51:16]: Checking rownames for catalog matrix and signature matrix.
## ✓ [2021-12-04 23:51:16]: Checked.
## ✓ [2021-12-04 23:51:16]: Method 'QP' detected.
## ✓ [2021-12-04 23:51:16]: Corresponding function generated.
## ℹ [2021-12-04 23:51:16]: Calling function.
## ℹ [2021-12-04 23:51:16]: Fitting sample: TCGA-A8-A09G-01A-21W-A019-09
## ✓ [2021-12-04 23:51:16]: Done.
## ℹ [2021-12-04 23:51:16]: Generating output signature exposures.
## ✓ [2021-12-04 23:51:16]: Done.
## ℹ [2021-12-04 23:51:16]: Calculating errors (Frobenius Norm).
## ✓ [2021-12-04 23:51:16]: Done.
## ℹ [2021-12-04 23:51:16]: 0.045 secs elapsed.
## ℹ [2021-12-04 23:51:16]: Getting bootstrap exposures (&errors/similarity) for different methods.
## ℹ [2021-12-04 23:51:16]: This step is time consuming, please be patient.
## ℹ [2021-12-04 23:51:16]: Processing sample `TCGA-A8-A09G-01A-21W-A019-09`.
## ✓ [2021-12-04 23:51:17]: Gotten.
## ℹ [2021-12-04 23:51:17]: Reporting p values...
## ℹ [2021-12-04 23:51:17]: Started.
## ✓ [2021-12-04 23:51:17]: Batch mode enabled.
## ✓ [2021-12-04 23:51:17]: Done.
## ℹ [2021-12-04 23:51:17]: 0.016 secs elapsed.
## ✓ [2021-12-04 23:51:17]: Done.
## ℹ [2021-12-04 23:51:17]: Cleaning results...
## ✓ [2021-12-04 23:51:17]: Outputing.
## ℹ [2021-12-04 23:51:17]: Total 1.181 secs elapsed.
bt_result
## $expo
## method sample sig exposure type
## 1: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 optimal
## 2: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.000000 optimal
## 3: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 357.000000 optimal
## 4: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.571227 Rep_1
## 5: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 11.191972 Rep_1
## 6: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 345.236801 Rep_1
## 7: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 10.758870 Rep_2
## 8: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.000000 Rep_2
## 9: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 346.241130 Rep_2
## 10: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 Rep_3
## 11: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 2.037155 Rep_3
## 12: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 354.962845 Rep_3
## 13: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 Rep_4
## 14: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.000000 Rep_4
## 15: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 357.000000 Rep_4
## 16: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.973130 Rep_5
## 17: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 2.350855 Rep_5
## 18: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 353.676014 Rep_5
## 19: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 5.262795 Rep_6
## 20: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 12.971145 Rep_6
## 21: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 338.766060 Rep_6
## 22: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 Rep_7
## 23: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.000000 Rep_7
## 24: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 357.000000 Rep_7
## 25: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.917790 Rep_8
## 26: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.384690 Rep_8
## 27: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 355.697520 Rep_8
## 28: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 Rep_9
## 29: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 13.629205 Rep_9
## 30: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 343.370795 Rep_9
## 31: QP TCGA-A8-A09G-01A-21W-A019-09 Sig1 0.000000 Rep_10
## 32: QP TCGA-A8-A09G-01A-21W-A019-09 Sig2 0.000000 Rep_10
## 33: QP TCGA-A8-A09G-01A-21W-A019-09 Sig3 357.000000 Rep_10
## method sample sig exposure type
##
## $error
## method sample errors type
## 1: QP TCGA-A8-A09G-01A-21W-A019-09 18.313 optimal
## 2: QP TCGA-A8-A09G-01A-21W-A019-09 18.930 Rep_1
## 3: QP TCGA-A8-A09G-01A-21W-A019-09 18.824 Rep_2
## 4: QP TCGA-A8-A09G-01A-21W-A019-09 18.353 Rep_3
## 5: QP TCGA-A8-A09G-01A-21W-A019-09 18.313 Rep_4
## 6: QP TCGA-A8-A09G-01A-21W-A019-09 18.392 Rep_5
## 7: QP TCGA-A8-A09G-01A-21W-A019-09 19.563 Rep_6
## 8: QP TCGA-A8-A09G-01A-21W-A019-09 18.313 Rep_7
## 9: QP TCGA-A8-A09G-01A-21W-A019-09 18.337 Rep_8
## 10: QP TCGA-A8-A09G-01A-21W-A019-09 19.122 Rep_9
## 11: QP TCGA-A8-A09G-01A-21W-A019-09 18.313 Rep_10
##
## $cosine
## method sample cosine type
## 1: QP TCGA-A8-A09G-01A-21W-A019-09 0.988618 optimal
## 2: QP TCGA-A8-A09G-01A-21W-A019-09 0.989479 Rep_1
## 3: QP TCGA-A8-A09G-01A-21W-A019-09 0.966835 Rep_2
## 4: QP TCGA-A8-A09G-01A-21W-A019-09 0.972369 Rep_3
## 5: QP TCGA-A8-A09G-01A-21W-A019-09 0.971626 Rep_4
## 6: QP TCGA-A8-A09G-01A-21W-A019-09 0.982418 Rep_5
## 7: QP TCGA-A8-A09G-01A-21W-A019-09 0.984565 Rep_6
## 8: QP TCGA-A8-A09G-01A-21W-A019-09 0.984212 Rep_7
## 9: QP TCGA-A8-A09G-01A-21W-A019-09 0.982291 Rep_8
## 10: QP TCGA-A8-A09G-01A-21W-A019-09 0.987838 Rep_9
## 11: QP TCGA-A8-A09G-01A-21W-A019-09 0.974672 Rep_10
##
## $p_val
## sample method threshold sig p_value
## 1: TCGA-A8-A09G-01A-21W-A019-09 QP 0.05 Sig1 9.999999e-01
## 2: TCGA-A8-A09G-01A-21W-A019-09 QP 0.05 Sig2 9.999783e-01
## 3: TCGA-A8-A09G-01A-21W-A019-09 QP 0.05 Sig3 4.978413e-17You can plot the result very easily with functions provided by sigminer.
show_sig_bootstrap_exposure(bt_result, sample = "TCGA-A8-A09G-01A-21W-A019-09")
## ℹ [2021-12-04 23:51:17]: Started.
## ℹ [2021-12-04 23:51:17]: Plotting.
## ℹ [2021-12-04 23:51:18]: 0.064 secs elapsed.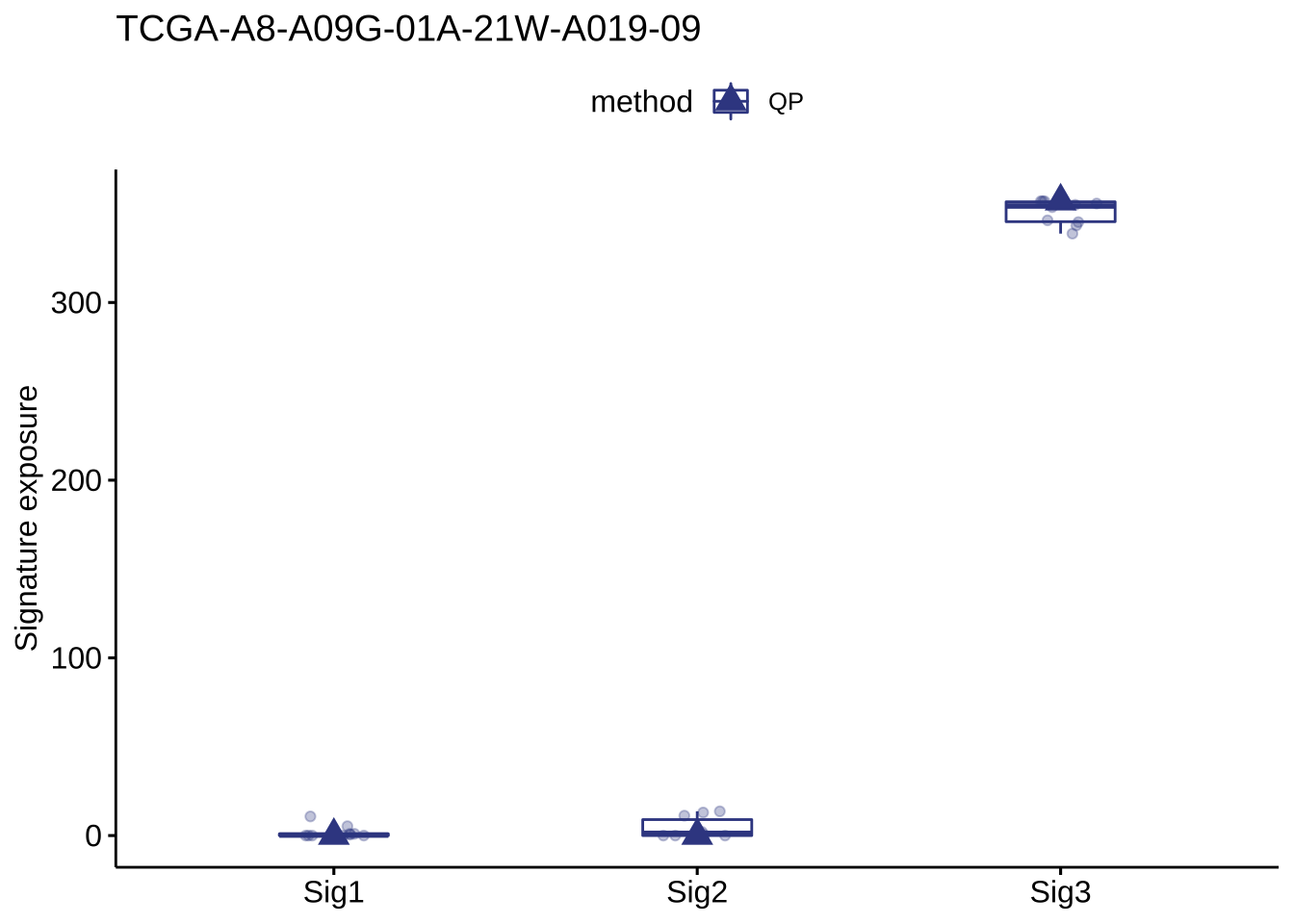
show_sig_bootstrap_error(bt_result, sample = "TCGA-A8-A09G-01A-21W-A019-09")
## ℹ [2021-12-04 23:51:18]: Started.
## ℹ [2021-12-04 23:51:18]: Plotting.
## ℹ [2021-12-04 23:51:18]: 0.043 secs elapsed.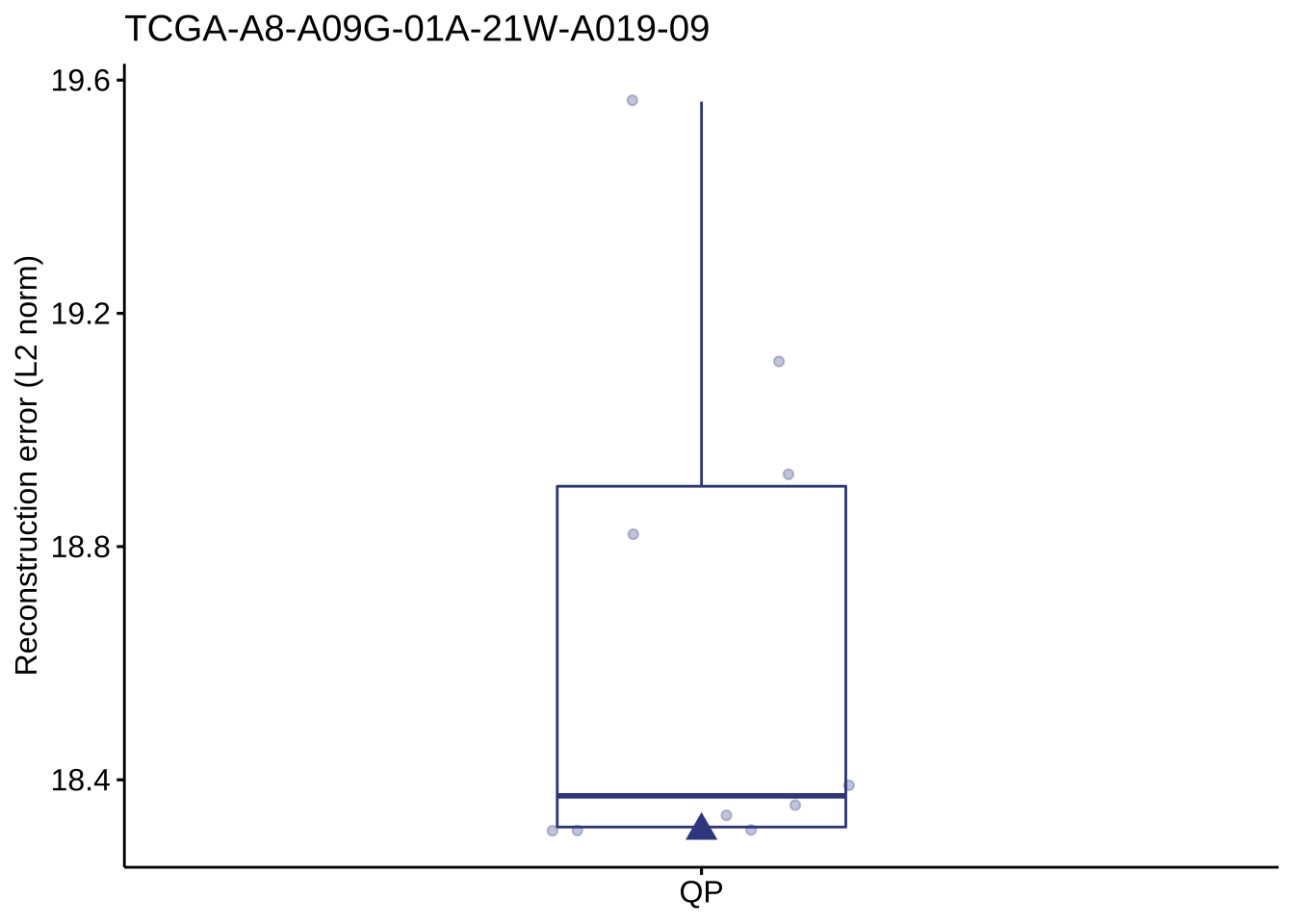
show_sig_bootstrap_stability(bt_result)
## ℹ [2021-12-04 23:51:18]: Started.
## ℹ [2021-12-04 23:51:18]: Plotting.
## ℹ [2021-12-04 23:51:18]: 0.037 secs elapsed.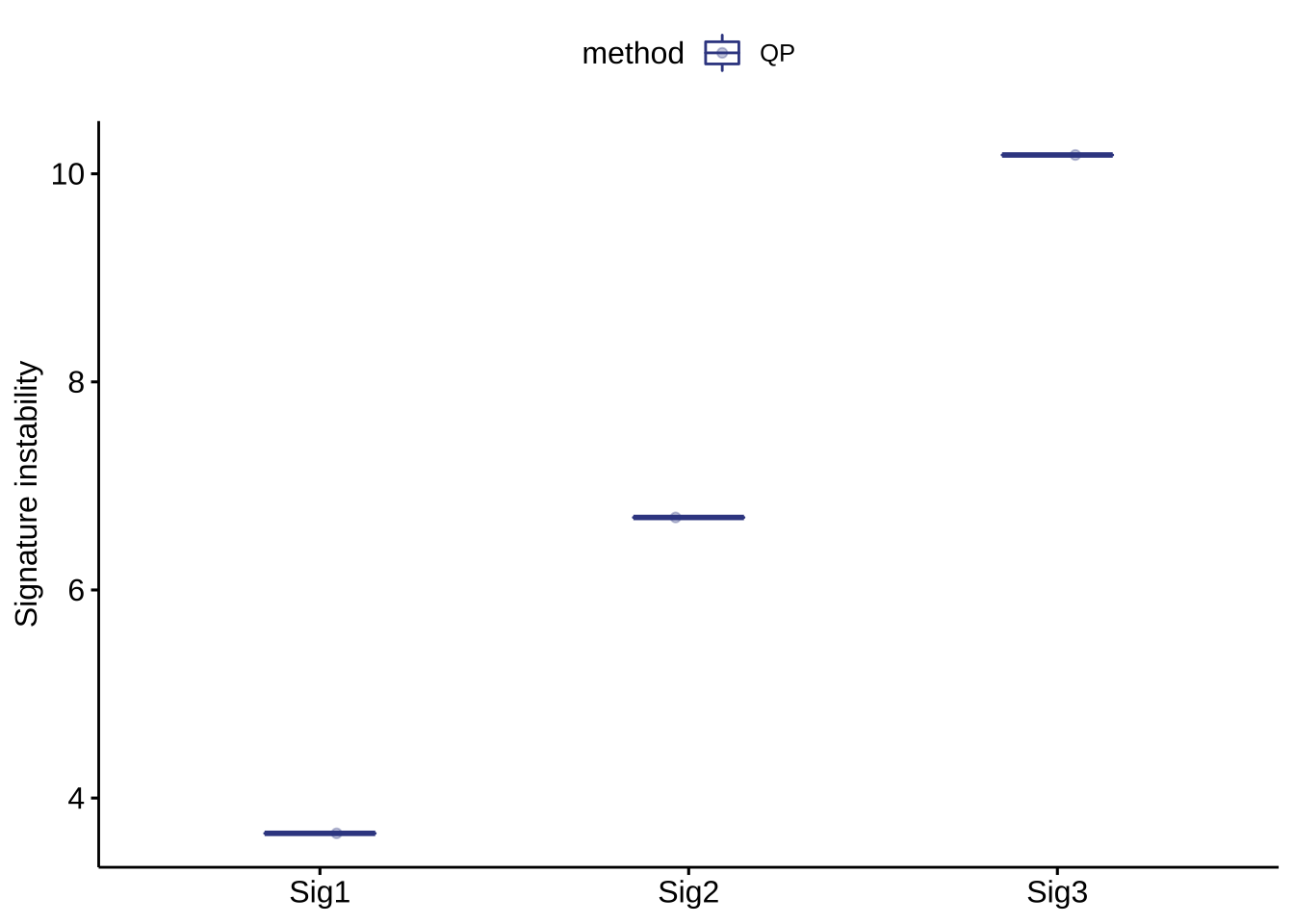
P values have been calculated under specified relative exposure cutoff (0.05 at default).