A Best Practice for Signature Extraction and Exposure (Activity) Attribution
Source:R/best_practice.R
bp.RdThese functions are combined to provide a best practice for optimally identifying mutational signatures and attributing their activities (exposures) in tumor samples. They are listed in order to use.
bp_extract_signatures()for extracting signatures.bp_show_survey()for showing measures change under different signature numbers to help user select optimal signature number. At default, an aggregated score (named score) is generated to suggest the best solution.bp_show_survey2()for showing simplified signature number survey likeshow_sig_number_survey().bp_get_sig_obj()for get a (list of)Signatureobject which is common used in sigminer for analysis and visualization.bp_attribute_activity()for optimizing signature activities (exposures). NOTE: the activities from extraction step may be better! You can also use sig_extract to get optimal NMF result from multiple NMF runs. Besides, you can use sig_fit to quantify exposures based on signatures extracted frombp_extract_signatures().bp_extract_signatures_iter()for extracting signature in a iteration way.bp_cluster_iter_list()for clustering (hclustwith average linkage) iterated signatures to help collapse multiple signatures into one. The result cluster can be visualized byplot()orfactoextra::fviz_dend().bp_get_clustered_sigs()for getting clustered (grouped) mean signatures from signature clusters.Extra:
bp_get_stats() for obtaining stats for signatures and samples of a solution. These stats are aggregated (averaged) as the stats for a solution (specific signature number).Extra:
bp_get_rank_score()for obtaining rank score for all signature numbers.
bp_extract_signatures(
nmf_matrix,
range = 2:5,
n_bootstrap = 20L,
n_nmf_run = 50,
RTOL = 0.001,
min_contribution = 0,
cores = parallelly::availableCores(max = 4),
cores_solution = min(cores, length(range)),
seed = 123456L,
handle_hyper_mutation = TRUE,
report_integer_exposure = FALSE,
only_core_stats = nrow(nmf_matrix) > 100,
cache_dir = file.path(tempdir(), "sigminer_bp"),
keep_cache = FALSE,
pynmf = FALSE,
use_conda = TRUE,
py_path = "/Users/wsx/anaconda3/bin/python"
)
bp_extract_signatures_iter(
nmf_matrix,
range = 2:5,
sim_threshold = 0.95,
max_iter = 10L,
n_bootstrap = 20L,
n_nmf_run = 50,
RTOL = 0.001,
min_contribution = 0,
cores = parallelly::availableCores(max = 4),
cores_solution = min(cores, length(range)),
seed = 123456L,
handle_hyper_mutation = TRUE,
report_integer_exposure = FALSE,
only_core_stats = nrow(nmf_matrix) > 100,
cache_dir = file.path(tempdir(), "sigminer_bp"),
keep_cache = FALSE,
pynmf = FALSE,
use_conda = FALSE,
py_path = "/Users/wsx/anaconda3/bin/python"
)
bp_cluster_iter_list(x, k = NULL, include_final_iteration = TRUE)
bp_get_clustered_sigs(SigClusters, cluster_label)
bp_get_sig_obj(obj, signum = NULL)
bp_get_stats(obj)
bp_get_rank_score(obj)
bp_show_survey2(
obj,
x = "signature_number",
left_y = "silhouette",
right_y = "L2_error",
left_name = left_y,
right_name = right_y,
left_color = "black",
right_color = "red",
left_shape = 16,
right_shape = 18,
shape_size = 4,
highlight = NULL
)
bp_show_survey(
obj,
add_score = FALSE,
scales = c("free_y", "free"),
fixed_ratio = TRUE
)
bp_attribute_activity(
input,
sample_class = NULL,
nmf_matrix = NULL,
method = c("bt", "stepwise"),
bt_use_prop = FALSE,
return_class = c("matrix", "data.table"),
use_parallel = FALSE,
cache_dir = file.path(tempdir(), "sigminer_attribute_activity"),
keep_cache = FALSE
)Arguments
- nmf_matrix
a
matrixused for NMF decomposition with rows indicate samples and columns indicate components.- range
a
numericvector containing the ranks of factorization to try. Note that duplicates are removed and values are sorted in increasing order. The results are notably returned in this order.- n_bootstrap
number of bootstrapped (resampling) catalogs used. When it is
0, the original (input) mutation catalog is used for NMF decomposition, this is not recommended, just for testing, user should not set it to0.- n_nmf_run
number of NMF runs for each bootstrapped or original catalog. At default, in total n_bootstrap x n_nmf_run (i.e. 1000) NMF runs are used for the task.
- RTOL
a threshold proposed by Nature Cancer paper to control how to filter solutions of NMF. Default is
0.1%(from reference #2), only NMF solutions with KLD (KL deviance) <=100.1%minimal KLD are kept.- min_contribution
a component contribution threshold to filer out small contributed components.
- cores
number of cpu cores to run NMF.
- cores_solution
cores for processing solutions, default is equal to argument
cores.- seed
a random seed to make reproducible result.
- handle_hyper_mutation
default is
TRUE, handle hyper-mutant samples.- report_integer_exposure
if
TRUE, report integer signature exposure by bootstrapping technique.- only_core_stats
if
TRUE, only calculate the core stats for signatures and samples.- cache_dir
a directory for keep temp result files.
- keep_cache
if
TRUE, keep cache results.- pynmf
if
TRUE, use Python NMF driver Nimfa. The seed currently is not used by this implementation, so the only way to reproduce your result is settingkeep_cache = TRUE.- use_conda
if
TRUE, create an independent conda environment to run NMF.- py_path
path to Python executable file, e.g. '/Users/wsx/anaconda3/bin/python'. In my test, it is more stable than
use_conda=TRUE. You can install the Nimfa package by yourself or setuse_condatoTRUEto install required Python environment, and then set this option.- sim_threshold
a similarity threshold for selecting samples to auto-rerun the extraction procedure (i.e.
bp_extract_signatures()), default is0.95.- max_iter
the maximum iteration size, default is 10, i.e., at most run the extraction procedure 10 times.
- x
result from
bp_extract_signatures_iter()or a list ofSignatureobjects.- k
an integer sequence specifying the cluster number to get silhouette.
- include_final_iteration
if
FALSE, exclude final iteration result from clustering for input frombp_extract_signatures_iter(), not applied if input is a list ofSignatureobjects.- SigClusters
result from
bp_cluster_iter_list().- cluster_label
cluster labels for a specified cluster number, obtain it from
SigClusters$sil_df.- obj
a
ExtractionResultobject frombp_extract_signatures().- signum
a integer vector to extract the corresponding
Signatureobject(s). If it isNULL(default), all will be returned.- left_y
column name for left y axis.
- right_y
column name for right y axis.
- left_name
label name for left y axis.
- right_name
label name for right y axis.
- left_color
color for left axis.
- right_color
color for right axis.
- left_shape, right_shape, shape_size
shape setting.
- highlight
a
integerto highlight ax.- add_score
if
FALSE, don't show score and label optimal points by rank score.- scales
one of "free_y" (default) and "free" to control the scales of plot facet.
- fixed_ratio
if
TRUE(default), make the x/y axis ratio fixed.- input
result from
bp_extract_signatures()or a Signature object.- sample_class
a named string vector whose names are sample names and values are class labels (i.e. cancer subtype). If it is
NULL(the default), treat all samples as one group.- method
one of 'bt' (use bootstrap exposure median, from reference #2, the most recommended way in my personal view) or stepwise' (stepwise reduce and update signatures then do signature fitting with last signature sets, from reference #2, the result tends to assign the contribution of removed signatures to the remaining signatures, maybe I misunderstand the paper method? PAY ATTENTION).
- bt_use_prop
this parameter is only used for
btmethod to reset low contributing signature activity (relative activity<0.01). IfTRUE, use empirical P value calculation way (i.e. proportion, used by reference#2), otherwise at.testis applied.- return_class
string, 'matrix' or 'data.table'.
- use_parallel
if
TRUE, use parallel computation based on furrr package. It can also be an integer for specifying cores.
Value
It depends on the called function.
Details
The signature extraction approach is adopted from reference #1, #2, and the whole best practice is adopted from the pipeline used by reference #3. I implement the whole procedure with R code based on the method description of papers. The code is well organized, tested and documented so user will find it pretty simple and useful. Besides, the structure of the results is very clear to see and also visualize like other approaches provided by sigminer.
Measure Explanation in Survey Plot
The survey plot provides a pretty good way to facilitate the signature number
selection. A score measure is calculated as the weighted mean of selected
measures and visualized as the first sub-plot. The optimal number is highlighted
with red color dot and the best values for each measures are also
highlighted with orange color dots. The detail of 6 measures shown in plot are
explained as below.
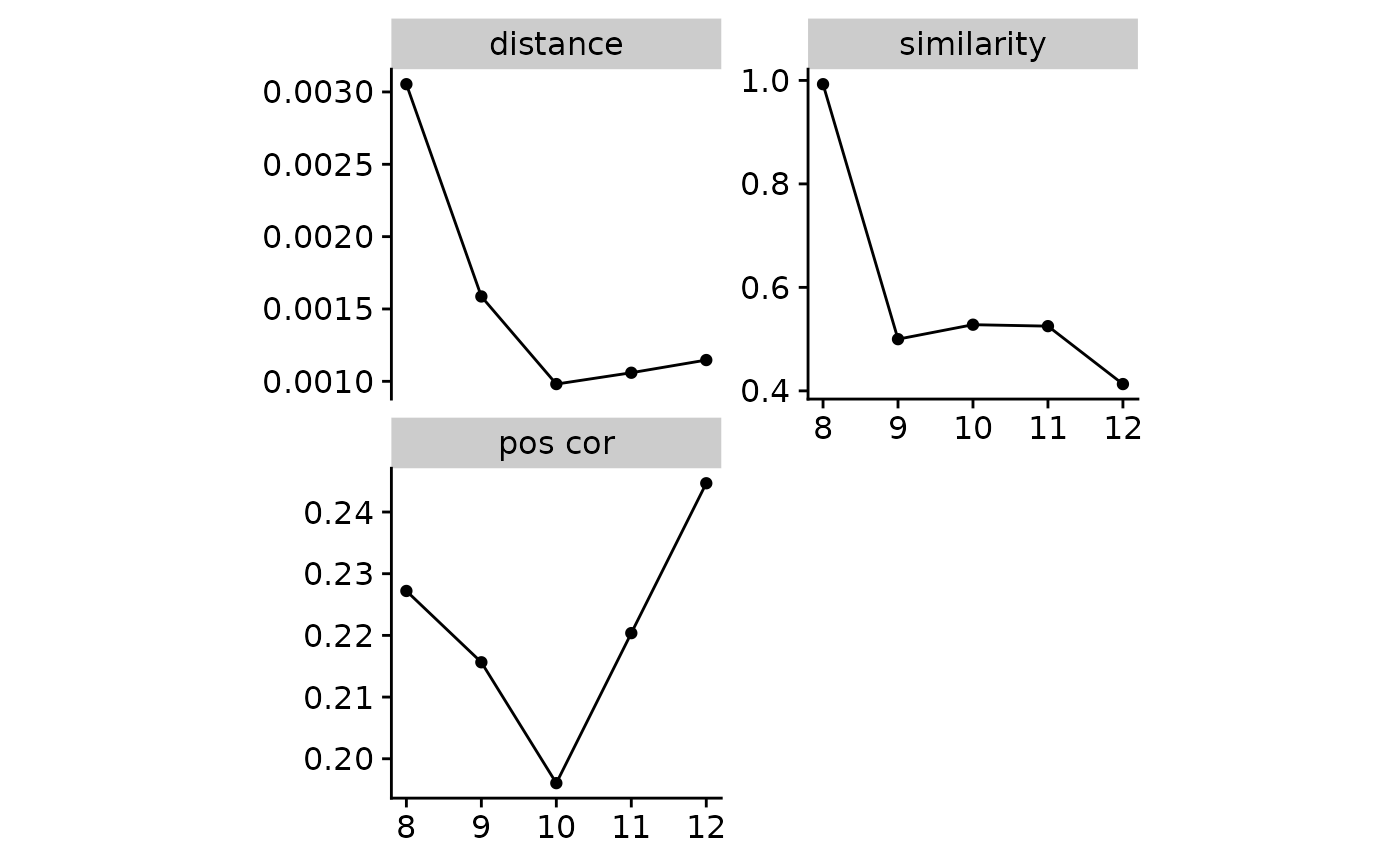
score- an aggregated score based on rank scores from selected measures below. The higher, the better. When two signature numbers have the same score, the larger signature number is preferred (this is a rare situation, you have to double check other measures).silhouette- the average silhouette width for signatures, also named as ASW in reference #2. The signature number with silhouette decreases sharply is preferred.distance- the average sample reconstructed cosine distance, the lower value is better.error- the average sample reconstructed error calculated with L2 formula (i.e. L2 error). This lower value is better. This measure represents a similar concept likedistanceabove, they are all used to quantify how well sample mutation profiles can be reconstructed from signatures, butdistancecares the whole mutation profile similarity whileerrorhere cares value difference.pos cor- the average positive signature exposure correlation coefficient. The lower value is better. This measure is constructed based on my understanding about signatures: mutational signatures are typically treated as independent recurrent patterns, so their activities are less correlated.similarity- the average similarity within in a signature cluster. Likesilhouette, the point decreases sharply is preferred. In the practice, results from multiple NMF runs are clustered with "clustering with match" algorithm proposed by reference #2. This value indicates if the signature profiles extracted from different NMF runs are similar.
References
Alexandrov, Ludmil B., et al. "Deciphering signatures of mutational processes operative in human cancer." Cell reports 3.1 (2013): 246-259.
Degasperi, Andrea, et al. "A practical framework and online tool for mutational signature analyses show intertissue variation and driver dependencies." Nature cancer 1.2 (2020): 249-263.
Alexandrov, Ludmil B., et al. “The repertoire of mutational signatures in human cancer.” Nature 578.7793 (2020): 94-101.
See also
See sig_estimate, sig_extract, sig_auto_extract, sigprofiler_extract for other approaches.
Examples
data("simulated_catalogs")
# \donttest{
# Here I reduce the values for n_bootstrap and n_nmf_run
# for reducing the run time.
# In practice, you should keep default or increase the values
# for better estimation.
#
# The input data here is simulated from 10 mutational signatures
# e1 <- bp_extract_signatures(
# t(simulated_catalogs$set1),
# range = 8:12,
# n_bootstrap = 5,
# n_nmf_run = 10
# )
#
# To avoid computation in examples,
# Here just load the result
# (e1$signature and e1$exposure set to NA to reduce package size)
load(system.file("extdata", "e1.RData", package = "sigminer"))
# See the survey for different signature numbers
# The suggested solution is marked as red dot
# with highest integrated score.
p1 <- bp_show_survey(e1)
p1
 # You can also exclude plotting and highlighting the score
p2 <- bp_show_survey(e1, add_score = FALSE)
p2
# You can also exclude plotting and highlighting the score
p2 <- bp_show_survey(e1, add_score = FALSE)
p2
 # You can also plot a simplified version
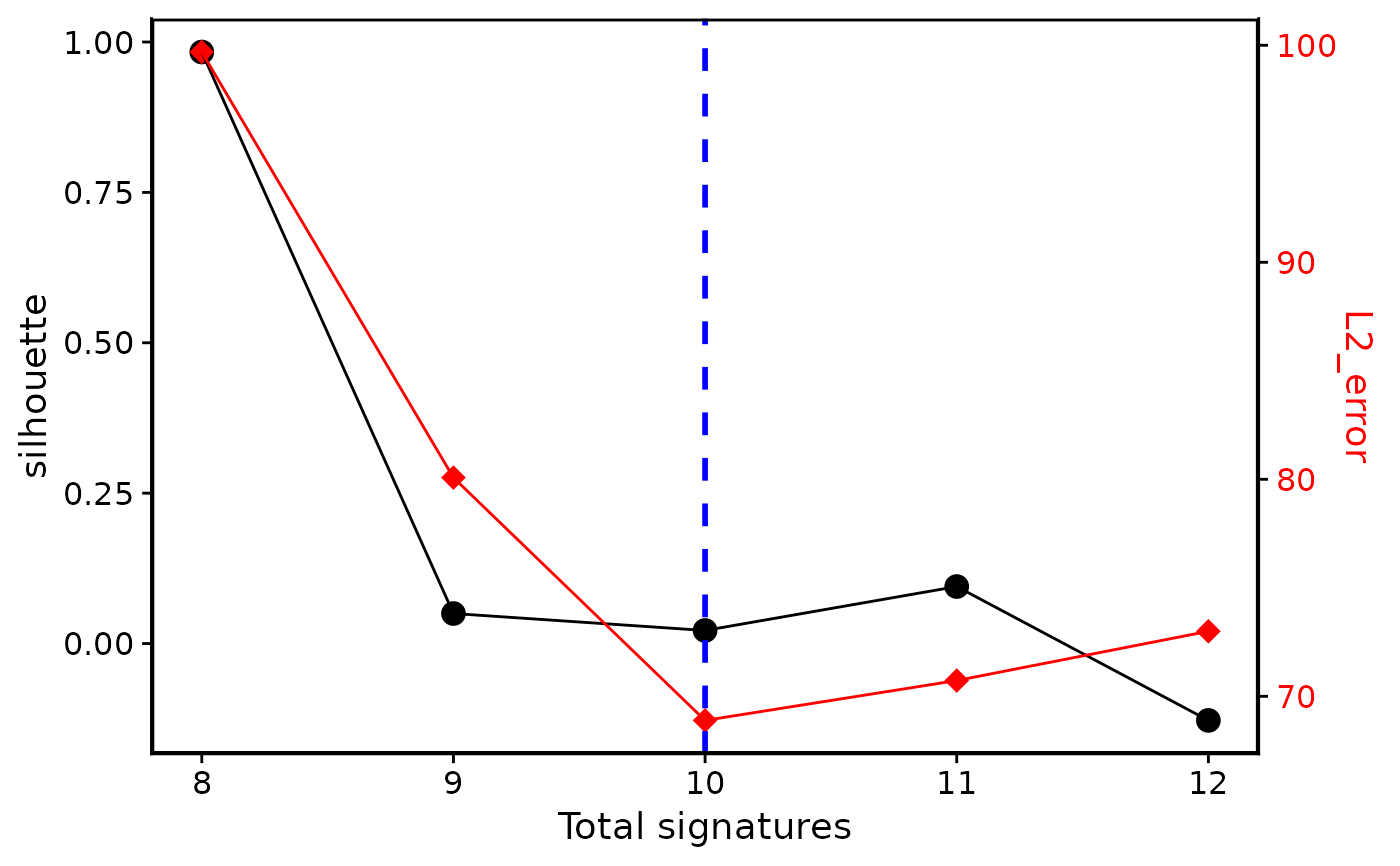
p3 <- bp_show_survey2(e1, highlight = 10)
#> Variables can be used: signature_number, silhouette, sample_cosine_distance, L1_error, L2_error, exposure_positive_correlation, signature_similarity_within_cluster, signature_similarity_across_cluster, silhouette_sample
#> Warning: The `size` argument of `element_rect()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> ℹ The deprecated feature was likely used in the sigminer package.
#> Please report the issue at <https://github.com/ShixiangWang/sigminer/issues>.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the sigminer package.
#> Please report the issue at <https://github.com/ShixiangWang/sigminer/issues>.
p3
# You can also plot a simplified version
p3 <- bp_show_survey2(e1, highlight = 10)
#> Variables can be used: signature_number, silhouette, sample_cosine_distance, L1_error, L2_error, exposure_positive_correlation, signature_similarity_within_cluster, signature_similarity_across_cluster, silhouette_sample
#> Warning: The `size` argument of `element_rect()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> ℹ The deprecated feature was likely used in the sigminer package.
#> Please report the issue at <https://github.com/ShixiangWang/sigminer/issues>.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the sigminer package.
#> Please report the issue at <https://github.com/ShixiangWang/sigminer/issues>.
p3
 # Obtain the suggested solution from extraction result
obj_suggested <- bp_get_sig_obj(e1, e1$suggested)
obj_suggested
#> $Signature
#> Sig1 Sig2 Sig3 Sig4 Sig5 Sig6
#> A[C>A]A 364.6855115 346.94762 452.99236 692.34882 380.33725 1.324947e+03
#> A[C>A]C 144.9048999 252.82263 388.79723 440.77833 227.80360 1.328586e+03
#> A[C>A]G 101.9965735 56.31129 73.55796 77.30450 35.81481 7.254115e+01
#> A[C>A]T 228.2381304 215.37118 321.71075 477.84538 175.07128 1.261005e+03
#> C[C>A]A 453.0228668 373.09497 354.26747 877.39212 132.35013 9.321536e+02
#> C[C>A]C 352.2870235 356.88211 261.08852 585.38882 103.33427 9.466691e+02
#> C[C>A]G 139.5844540 77.21084 71.37286 89.08911 67.52359 5.770273e+00
#> C[C>A]T 206.6452684 325.06644 233.99811 449.18696 224.99675 9.577429e+02
#> G[C>A]A 262.9030436 93.42677 249.76023 671.83989 292.79971 6.752285e+02
#> G[C>A]C 234.4691391 203.94031 338.46614 341.08226 239.81114 5.328750e+02
#> G[C>A]G 77.2748163 38.76242 67.73828 55.54570 33.87816 2.079253e+01
#> G[C>A]T 25.1498022 147.95376 306.35190 471.88666 250.35555 6.924583e+02
#> T[C>A]A 707.9694364 1128.97194 915.45972 578.90913 425.73686 9.433380e+02
#> T[C>A]C 550.3665329 639.22968 560.35636 691.10963 363.64965 5.479057e+02
#> T[C>A]G 124.1977001 90.18331 156.15108 117.38406 93.80205 1.223391e+01
#> T[C>A]T 423.1960918 487.78631 622.24017 648.89415 477.28252 1.218222e+03
#> A[C>G]A 191.6864937 322.45523 599.97352 496.63443 220.62923 3.340177e+02
#> A[C>G]C 98.4509868 163.72693 305.70556 240.58008 192.32289 1.778502e+02
#> A[C>G]G 32.9514639 41.90439 110.29973 130.08351 51.99749 5.024744e-06
#> A[C>G]T 196.5392614 354.14978 543.65236 573.08529 259.68447 2.734687e+02
#> C[C>G]A 170.7588970 286.52200 405.42625 425.08854 151.77176 2.007334e+02
#> C[C>G]C 184.4906424 173.75603 306.77347 324.29104 135.08277 1.839090e+02
#> C[C>G]G 37.1196263 34.60268 108.73587 98.87709 47.68770 2.160016e+01
#> C[C>G]T 191.1959730 337.55583 526.49460 653.67282 177.24473 2.172862e+02
#> G[C>G]A 120.8305851 147.81819 245.41235 278.24330 94.40144 1.447409e+02
#> G[C>G]C 85.9454946 129.07501 248.42925 244.06828 203.54229 1.119571e+02
#> G[C>G]G 11.2368880 18.09011 48.93059 39.78653 18.81565 1.239872e+00
#> G[C>G]T 99.2539804 194.17868 318.01486 404.69066 119.83849 2.243552e+02
#> T[C>G]A 2304.8190218 5806.53390 2543.20013 827.06428 4.47700 8.045533e+01
#> T[C>G]C 649.4320013 1498.46533 908.51263 607.96705 148.26065 5.881513e+01
#> T[C>G]G 91.1666377 209.73580 147.99082 71.38912 23.82618 8.602967e-04
#> T[C>G]T 2761.4204889 6844.53103 3311.67302 1324.25369 108.29443 6.392960e+01
#> A[C>T]A 580.6367660 486.55883 771.14239 573.23727 1045.30267 8.209856e+02
#> A[C>T]C 336.8462426 166.00633 384.82160 259.72339 579.17288 2.279499e+02
#> A[C>T]G 41.2744554 48.63027 1095.73657 279.15822 5056.19068 7.232503e+02
#> A[C>T]T 365.5627960 320.98049 532.98918 433.35276 478.77227 7.802344e+02
#> C[C>T]A 1107.9997769 578.99821 714.60791 489.10670 828.90566 1.504848e+02
#> C[C>T]C 680.5296357 284.55116 583.37633 291.95102 705.17673 1.096355e+02
#> C[C>T]G 140.6997922 110.49878 608.69038 382.82184 2856.59934 9.769086e+01
#> C[C>T]T 899.7884560 535.31877 570.95299 596.89330 769.16400 7.430400e+02
#> G[C>T]A 740.5029427 445.50554 653.16343 432.39515 1022.83916 4.543718e+01
#> G[C>T]C 518.2147424 336.90086 473.33371 288.04137 1073.90995 8.017087e-01
#> G[C>T]G 151.0536663 97.06964 535.59679 111.37791 3150.67238 3.028044e+02
#> G[C>T]T 684.1239448 427.63207 522.89528 260.30904 744.80860 2.399426e+02
#> T[C>T]A 1935.5135649 2371.00890 1254.10598 184.58238 468.56327 2.016729e+02
#> T[C>T]C 808.7068291 534.84839 784.12753 396.90968 736.96715 7.805433e+01
#> T[C>T]G 174.8872796 75.86828 502.79237 130.24016 2179.16906 1.917718e+02
#> T[C>T]T 887.6696088 710.46176 664.34758 223.84417 452.25297 3.454048e+02
#> A[T>A]A 317.6552554 228.37936 246.26228 271.09229 168.31318 4.928755e+02
#> A[T>A]C 278.2529058 213.13425 260.76926 232.40386 98.31839 4.406474e+02
#> A[T>A]G 249.1917651 182.23415 340.62694 225.47731 185.96380 5.335190e+02
#> A[T>A]T 246.2567101 310.95073 335.99184 190.22561 288.31773 1.114181e+03
#> C[T>A]A 100.2452810 96.44490 181.15835 160.06344 66.32999 5.002986e+02
#> C[T>A]C 315.3312658 290.19171 210.47003 393.73526 34.30914 7.272778e+02
#> C[T>A]G 214.4139116 134.29338 313.33861 255.20058 152.95544 6.791202e+02
#> C[T>A]T 255.1055329 189.25835 347.36489 510.57007 130.67930 9.858692e+02
#> G[T>A]A 78.5708695 46.54815 181.40288 119.84166 92.06006 3.475210e+02
#> G[T>A]C 162.1735392 152.29781 122.00953 162.13346 53.23410 2.791327e+02
#> G[T>A]G 159.3889064 110.89872 247.32946 192.69463 94.97434 3.082144e+02
#> G[T>A]T 115.6398251 140.47073 153.38354 297.73054 26.48535 7.054885e+02
#> T[T>A]A 252.4670358 160.68865 305.17529 216.26710 228.23242 6.535623e+02
#> T[T>A]C 592.0449289 353.72488 228.79198 337.76826 55.16387 4.069120e+02
#> T[T>A]G 198.0755104 151.91335 256.84425 226.22452 62.18204 3.285117e+02
#> T[T>A]T 383.5737823 337.00589 351.72460 404.10447 88.98538 1.036281e+03
#> A[T>C]A 2214.2964522 1259.06003 851.26760 769.08597 645.92386 6.571294e+02
#> A[T>C]C 1296.4130889 789.71635 429.95798 612.10931 168.38083 3.339987e+02
#> A[T>C]G 2184.7595181 1231.70050 712.42662 866.79096 423.38503 3.059365e+02
#> A[T>C]T 1334.2884205 746.77707 823.13917 536.52760 381.41889 9.212770e+02
#> C[T>C]A 1368.4259217 768.78881 352.44770 550.53435 178.86216 1.540993e+02
#> C[T>C]C 1517.2716800 984.20451 361.66402 904.29318 97.95732 2.679858e+02
#> C[T>C]G 1890.4159043 1020.51180 435.96306 693.79180 283.74277 7.832913e+01
#> C[T>C]T 1419.4825104 789.41692 461.84585 894.63908 253.60587 3.501025e+02
#> G[T>C]A 2449.4241104 1446.44266 424.35425 906.38195 218.56288 1.170186e+02
#> G[T>C]C 1114.6078783 664.36882 219.30814 553.12788 124.96875 2.110529e+02
#> G[T>C]G 1737.5192128 1021.54590 349.01345 654.90212 130.34669 1.528999e+02
#> G[T>C]T 1758.5622876 1017.16814 477.82218 719.38885 245.19309 1.661486e+02
#> T[T>C]A 2185.6729708 1226.57228 480.33544 787.78432 208.84153 1.190451e+02
#> T[T>C]C 1490.7693030 863.31094 418.77913 676.08017 163.41359 1.798135e+02
#> T[T>C]G 1357.3556066 889.25467 370.53252 521.65067 232.69739 2.304677e+01
#> T[T>C]T 1684.7172024 939.72966 463.64985 665.37852 175.01269 3.423028e+02
#> A[T>G]A 94.4330523 63.49243 100.69104 64.50282 68.14202 1.959488e+02
#> A[T>G]C 30.4353615 40.74580 67.32443 66.49769 63.33256 2.948087e+01
#> A[T>G]G 83.6873843 128.33196 167.15785 235.83434 149.35654 2.825874e+02
#> A[T>G]T 127.1360117 105.47819 139.83909 220.61187 114.86815 7.451436e+01
#> C[T>G]A 70.5662971 29.27212 72.12039 57.89206 24.15968 1.131150e+02
#> C[T>G]C 210.8441666 150.24265 126.53264 252.93096 93.03145 6.693238e+01
#> C[T>G]G 232.3968048 173.22663 216.46656 294.16258 156.93625 2.150109e+02
#> C[T>G]T 150.3588432 66.59398 208.90862 985.23861 129.16472 6.238572e+01
#> G[T>G]A 17.0229495 29.80699 61.77359 90.96154 31.34418 1.800350e+01
#> G[T>G]C 0.8591066 27.85169 43.31172 134.02653 11.43404 5.874917e+00
#> G[T>G]G 69.6919626 179.41070 333.42242 428.18415 107.86656 1.859646e+02
#> G[T>G]T 105.2678056 98.08502 80.19119 381.34504 43.04977 1.704416e+02
#> T[T>G]A 121.6433012 111.73067 186.94694 146.40000 97.89894 5.773345e+01
#> T[T>G]C 149.5333411 116.51460 196.94210 192.20642 108.00836 8.684462e+01
#> T[T>G]G 92.4731954 109.25682 245.52028 297.37939 121.43337 1.981042e+02
#> T[T>G]T 154.2386196 137.23642 463.48903 453.32162 362.20483 3.207298e+02
#> Sig7 Sig8 Sig9 Sig10 Sig11
#> A[C>A]A 1.612108e+03 513.18466 370.426103 317.485977 301.03623
#> A[C>A]C 2.811969e+02 401.26021 92.737155 172.921734 284.52485
#> A[C>A]G 6.406936e+01 34.86885 21.922641 40.546934 54.23260
#> A[C>A]T 5.420044e+02 442.76085 179.626853 174.936185 239.09711
#> C[C>A]A 2.982074e+03 378.20672 661.929329 410.372295 352.30301
#> C[C>A]C 6.180518e+02 407.35946 144.713698 496.039327 282.01916
#> C[C>A]G 5.695139e+02 70.25062 137.061730 181.714287 84.90837
#> C[C>A]T 1.145804e+03 595.39775 242.674959 1254.882610 543.60381
#> G[C>A]A 4.307915e+03 201.90842 1045.442740 335.581323 189.36150
#> G[C>A]C 4.907381e+02 281.13290 147.213294 218.435173 218.83653
#> G[C>A]G 3.010680e+02 50.90646 93.143500 75.067089 27.98970
#> G[C>A]T 2.381946e+03 311.50351 598.580470 307.131262 223.18275
#> T[C>A]A 2.260020e+03 184.81599 809.740232 341.930871 351.10781
#> T[C>A]C 1.420839e+03 272.18579 422.915978 265.454149 331.98760
#> T[C>A]G 4.926154e+02 61.94453 155.534013 38.590677 48.86329
#> T[C>A]T 3.949891e+03 485.04643 1038.476495 528.723002 445.57556
#> A[C>G]A 3.520491e+01 422.58229 40.719413 213.294440 268.63571
#> A[C>G]C 7.537606e+01 226.12183 61.341091 110.981307 143.04307
#> A[C>G]G 8.863639e+01 78.61895 22.544953 51.641726 43.82139
#> A[C>G]T 9.105450e+01 366.43408 89.752984 211.646507 271.03210
#> C[C>G]A 1.972337e+01 248.53555 60.458058 88.425799 197.55635
#> C[C>G]C 9.389712e+01 260.05125 47.392917 117.524661 142.99773
#> C[C>G]G 1.663936e+02 53.56070 52.251455 32.427322 48.24376
#> C[C>G]T 1.706537e+02 393.61001 110.455586 149.326789 252.83704
#> G[C>G]A 2.172857e+01 172.11493 40.207808 83.289982 144.21200
#> G[C>G]C 4.494280e+00 178.13304 24.373164 102.997208 79.65155
#> G[C>G]G 1.405500e+01 24.57657 8.070140 9.880719 23.98459
#> G[C>G]T 4.908029e+01 278.74457 48.651901 116.891518 199.79989
#> T[C>G]A 1.165248e+02 174.16061 2103.156522 60.437995 636.17041
#> T[C>G]C 2.559735e+02 319.73340 537.274027 144.375587 254.02401
#> T[C>G]G 4.842660e+01 36.11222 90.435720 21.618406 42.63096
#> T[C>G]T 3.079238e+02 437.76949 2451.564561 107.446794 1076.02647
#> A[C>T]A 8.516367e+01 509.78431 164.680745 682.596751 368.57769
#> A[C>T]C 6.549977e+01 275.39037 93.548518 407.421365 194.61958
#> A[C>T]G 1.681590e+02 639.81418 192.171636 848.451511 219.49651
#> A[C>T]T 1.133526e+02 443.09742 116.301412 436.498259 353.52505
#> C[C>T]A 4.266475e+02 486.79212 382.866038 341.456923 281.63431
#> C[C>T]C 1.837298e+02 297.96180 93.103677 314.845399 149.63889
#> C[C>T]G 4.588847e+02 656.23660 223.766036 1160.824899 367.12010
#> C[C>T]T 2.099212e+02 473.19977 195.353006 375.259764 216.10874
#> G[C>T]A 4.583719e+01 634.10065 70.067798 1225.877730 446.60656
#> G[C>T]C 5.966043e+01 555.87383 11.390655 1446.161268 410.37815
#> G[C>T]G 1.952663e+02 865.03568 150.635721 1883.402702 485.25151
#> G[C>T]T 5.219121e+01 482.18963 15.358399 1077.792737 381.75646
#> T[C>T]A 1.546390e+03 1274.21386 867.813705 186.347428 3610.62119
#> T[C>T]C 5.496995e+02 581.97699 331.764668 389.736658 828.37489
#> T[C>T]G 4.305419e+02 429.02041 299.813014 416.334741 431.72765
#> T[C>T]T 7.084075e+02 938.68885 305.542684 154.768464 2052.27825
#> A[T>A]A 7.306582e+01 214.07703 40.380191 176.207375 120.52509
#> A[T>A]C 2.160929e+01 184.66354 35.452354 183.943184 164.97223
#> A[T>A]G 1.005536e+02 223.40107 57.061578 153.156016 158.48139
#> A[T>A]T 5.196649e+00 178.04216 72.976932 225.995616 198.19835
#> C[T>A]A 1.031591e+01 178.96767 4.101657 69.662794 91.91138
#> C[T>A]C 7.133643e+01 258.40038 146.588241 183.514312 188.51210
#> C[T>A]G 1.508386e+02 241.73351 104.706117 199.029485 186.53684
#> C[T>A]T 5.950672e+01 450.47718 448.413420 159.310090 200.94527
#> G[T>A]A 4.023947e+01 132.03257 15.449153 135.074786 84.25939
#> G[T>A]C 4.059007e+01 144.93827 41.140153 122.359928 73.99045
#> G[T>A]G 2.666966e+01 206.97926 58.077096 108.049975 95.89027
#> G[T>A]T 1.039028e+02 162.86845 96.512208 139.668975 110.51575
#> T[T>A]A 1.479951e+02 177.40016 59.351331 101.861911 83.66533
#> T[T>A]C 3.988944e+01 221.61403 76.704589 99.678627 156.37407
#> T[T>A]G 4.386895e+01 131.16375 13.215947 112.645471 70.52114
#> T[T>A]T 9.197887e+01 404.64818 62.262232 159.609253 246.15917
#> A[T>C]A 1.191110e+02 335.01932 65.429821 530.206594 227.30323
#> A[T>C]C 3.904971e+01 159.92207 171.022183 136.603821 200.82597
#> A[T>C]G 1.352414e+02 311.18862 113.133780 613.917569 265.01951
#> A[T>C]T 1.187246e+02 354.08441 101.227039 455.991338 259.04444
#> C[T>C]A 3.547481e+01 178.23633 131.679803 173.902849 159.88132
#> C[T>C]C 1.181216e+02 353.55559 565.016939 165.869156 199.39352
#> C[T>C]G 8.349381e+01 349.37968 411.872251 602.032977 266.30015
#> C[T>C]T 9.314132e+01 515.71853 1405.308433 205.208256 138.86411
#> G[T>C]A 2.096227e+01 129.66327 24.431083 236.377912 202.21432
#> G[T>C]C 8.448178e+01 136.57622 155.816136 185.699517 116.65866
#> G[T>C]G 2.165845e+02 111.21861 116.556976 262.845807 110.23349
#> G[T>C]T 1.712354e+02 184.11356 153.641728 162.856944 144.86920
#> T[T>C]A 3.227932e+01 142.89343 39.611581 187.313337 144.31734
#> T[T>C]C 2.064432e+02 238.07017 205.195362 115.366520 204.09146
#> T[T>C]G 4.401696e+01 96.63450 113.903142 192.806438 146.76799
#> T[T>C]T 4.759346e+01 169.16982 133.755945 196.920385 184.60302
#> A[T>G]A 4.033890e+00 40.36780 11.242104 66.646853 27.33252
#> A[T>G]C 6.087864e+01 48.52501 106.739107 55.313219 25.22731
#> A[T>G]G 5.545958e+01 158.65964 72.653213 85.767824 107.17091
#> A[T>G]T 7.122083e+01 237.06369 438.170763 63.674406 78.92264
#> C[T>G]A 1.443062e-12 44.97803 7.262841 16.753545 22.04673
#> C[T>G]C 5.585688e+01 169.31173 273.464660 81.668878 73.01694
#> C[T>G]G 1.619524e+02 222.54191 236.659074 181.248153 202.89875
#> C[T>G]T 1.808814e+02 1014.28261 3612.855255 162.959024 11.20453
#> G[T>G]A 5.097588e+01 67.23368 15.831875 26.673109 40.87270
#> G[T>G]C 1.208215e+00 88.11666 129.743115 32.569949 57.50406
#> G[T>G]G 3.286240e+02 240.69052 135.338848 109.371542 165.10136
#> G[T>G]T 1.089361e+02 326.61030 840.187207 82.431326 82.28797
#> T[T>G]A 9.813250e+00 97.48130 11.504350 83.856599 71.65787
#> T[T>G]C 9.185830e+01 123.56889 152.977572 86.260669 107.28512
#> T[T>G]G 3.350509e+01 226.00057 86.625452 88.302890 110.06536
#> T[T>G]T 3.225139e+01 521.83563 761.382232 264.138090 175.67229
#>
#> $Signature.norm
#> Sig1 Sig2 Sig3 Sig4 Sig5
#> A[C>A]A 6.714744e-03 0.0070443097 0.011013137 0.017684877 0.0108444883
#> A[C>A]C 2.668050e-03 0.0051332270 0.009452426 0.011258936 0.0064953234
#> A[C>A]G 1.878004e-03 0.0011433259 0.001788339 0.001974612 0.0010211813
#> A[C>A]T 4.202417e-03 0.0043728252 0.007821422 0.012205750 0.0049917763
#> C[C>A]A 8.341249e-03 0.0075751968 0.008612940 0.022411495 0.0037736757
#> C[C>A]C 6.486458e-03 0.0072460163 0.006347576 0.014952765 0.0029463516
#> C[C>A]G 2.570088e-03 0.0015676634 0.001735215 0.002275630 0.0019252882
#> C[C>A]T 3.804840e-03 0.0066000415 0.005688955 0.011473720 0.0064152924
#> G[C>A]A 4.840682e-03 0.0018969062 0.006072163 0.017161012 0.0083485461
#> G[C>A]C 4.317145e-03 0.0041407367 0.008228778 0.008712368 0.0068376923
#> G[C>A]G 1.422817e-03 0.0007870193 0.001646851 0.001418821 0.0009659619
#> G[C>A]T 4.630688e-04 0.0030040043 0.007448018 0.012053545 0.0071383433
#> T[C>A]A 1.303543e-02 0.0229222728 0.022256629 0.014787253 0.0121389593
#> T[C>A]C 1.013358e-02 0.0129787080 0.013623367 0.017653224 0.0103686778
#> T[C>A]G 2.286781e-03 0.0018310520 0.003796340 0.002998377 0.0026745613
#> T[C>A]T 7.792065e-03 0.0099038519 0.015127884 0.016574901 0.0136086717
#> A[C>G]A 3.529413e-03 0.0065470243 0.014586538 0.012685685 0.0062907618
#> A[C>G]C 1.812721e-03 0.0033242574 0.007432304 0.006145210 0.0054836684
#> A[C>G]G 6.067163e-04 0.0008508129 0.002681604 0.003322763 0.0014825952
#> A[C>G]T 3.618764e-03 0.0071905401 0.013217260 0.014638493 0.0074043371
#> C[C>G]A 3.144085e-03 0.0058174479 0.009856711 0.010858166 0.0043274412
#> C[C>G]C 3.396920e-03 0.0035278849 0.007458267 0.008283465 0.0038515909
#> C[C>G]G 6.834623e-04 0.0007025615 0.002643583 0.002525648 0.0013597108
#> C[C>G]T 3.520381e-03 0.0068536218 0.012800121 0.016696964 0.0050537474
#> G[C>G]A 2.224784e-03 0.0030012516 0.005966458 0.007107254 0.0026916513
#> G[C>G]C 1.582465e-03 0.0026206963 0.006039804 0.006234311 0.0058035652
#> G[C>G]G 2.068983e-04 0.0003672956 0.001189599 0.001016280 0.0005364872
#> G[C>G]T 1.827506e-03 0.0039425397 0.007731568 0.010337137 0.0034169337
#> T[C>G]A 4.243730e-02 0.1178939434 0.061830204 0.021125956 0.0001276519
#> T[C>G]C 1.195762e-02 0.0304243444 0.022087731 0.015529488 0.0042273295
#> T[C>G]G 1.678599e-03 0.0042584064 0.003597948 0.001823514 0.0006793516
#> T[C>G]T 5.084444e-02 0.1389690939 0.080513294 0.033825817 0.0030877799
#> A[C>T]A 1.069093e-02 0.0098789295 0.018747990 0.014642375 0.0298045291
#> A[C>T]C 6.202155e-03 0.0033705377 0.009355771 0.006634194 0.0165138533
#> A[C>T]G 7.599627e-04 0.0009873728 0.026639514 0.007130624 0.1441662650
#> A[C>T]T 6.730897e-03 0.0065170816 0.012958017 0.011069262 0.0136511486
#> C[C>T]A 2.040096e-02 0.0117557881 0.017373526 0.012493402 0.0236344396
#> C[C>T]C 1.253020e-02 0.0057774325 0.014183028 0.007457394 0.0201065787
#> C[C>T]G 2.590624e-03 0.0022435307 0.014798462 0.009778535 0.0814497087
#> C[C>T]T 1.656729e-02 0.0108689351 0.013880992 0.015246628 0.0219310362
#> G[C>T]A 1.363445e-02 0.0090453971 0.015879689 0.011044801 0.0291640310
#> G[C>T]C 9.541589e-03 0.0068403236 0.011507675 0.007357529 0.0306202032
#> G[C>T]G 2.781264e-03 0.0019708699 0.013021413 0.002844960 0.0898345608
#> G[C>T]T 1.259638e-02 0.0086825002 0.012712614 0.006649153 0.0212365951
#> T[C>T]A 3.563749e-02 0.0481401803 0.030489786 0.004714844 0.0133600610
#> T[C>T]C 1.489025e-02 0.0108593848 0.019063685 0.010138385 0.0210130133
#> T[C>T]G 3.220099e-03 0.0015404044 0.012223873 0.003326764 0.0621342595
#> T[C>T]T 1.634415e-02 0.0144249805 0.016151598 0.005717720 0.0128950083
#> A[T>A]A 5.848803e-03 0.0046369389 0.005987121 0.006924593 0.0047990836
#> A[T>A]C 5.123310e-03 0.0043274074 0.006339814 0.005936363 0.0028033349
#> A[T>A]G 4.588224e-03 0.0037000218 0.008281312 0.005759436 0.0053023528
#> A[T>A]T 4.534183e-03 0.0063134407 0.008168623 0.004858991 0.0082207520
#> C[T>A]A 1.845759e-03 0.0019581853 0.004404316 0.004088549 0.0018912552
#> C[T>A]C 5.806013e-03 0.0058919565 0.005116941 0.010057300 0.0009782504
#> C[T>A]G 3.947880e-03 0.0027266485 0.007617879 0.006518666 0.0043611914
#> C[T>A]T 4.697111e-03 0.0038426390 0.008445125 0.013041647 0.0037260357
#> G[T>A]A 1.446680e-03 0.0009450981 0.004410261 0.003061152 0.0026248922
#> G[T>A]C 2.986008e-03 0.0030922044 0.002966292 0.004141425 0.0015178545
#> G[T>A]G 2.934736e-03 0.0022516508 0.006013066 0.004922058 0.0027079864
#> G[T>A]T 2.129209e-03 0.0028520713 0.003729056 0.007605022 0.0007551720
#> T[T>A]A 4.648530e-03 0.0032625691 0.007419412 0.005524177 0.0065075505
#> T[T>A]C 1.090098e-02 0.0071819130 0.005562384 0.008627718 0.0015728777
#> T[T>A]G 3.647050e-03 0.0030843984 0.006244390 0.005778522 0.0017729856
#> T[T>A]T 7.062523e-03 0.0068424561 0.008551118 0.010322164 0.0025372244
#> A[T>C]A 4.077056e-02 0.0255635383 0.020695992 0.019644998 0.0184171121
#> A[T>C]C 2.387011e-02 0.0160341395 0.010453125 0.015635295 0.0048010126
#> A[T>C]G 4.022672e-02 0.0250080395 0.017320494 0.022140706 0.0120719020
#> A[T>C]T 2.456748e-02 0.0151623147 0.020012135 0.013704688 0.0108753289
#> C[T>C]A 2.519604e-02 0.0156092336 0.008568698 0.014062467 0.0050998648
#> C[T>C]C 2.793665e-02 0.0199829629 0.008792765 0.023098637 0.0027930397
#> C[T>C]G 3.480714e-02 0.0207201340 0.010599121 0.017721736 0.0080903070
#> C[T>C]T 2.613611e-02 0.0160280599 0.011228382 0.022852039 0.0072310190
#> G[T>C]A 4.509983e-02 0.0293680932 0.010316888 0.023151991 0.0062318446
#> G[T>C]C 2.052263e-02 0.0134891247 0.005331813 0.014128714 0.0035632117
#> G[T>C]G 3.199194e-02 0.0207411301 0.008485204 0.016728365 0.0037165519
#> G[T>C]T 3.237939e-02 0.0206522455 0.011616798 0.018375569 0.0069911470
#> T[T>C]A 4.024354e-02 0.0249039178 0.011677901 0.020122616 0.0059546613
#> T[T>C]C 2.744867e-02 0.0175283797 0.010181346 0.017269322 0.0046593827
#> T[T>C]G 2.499221e-02 0.0180551327 0.009008375 0.013324682 0.0066348593
#> T[T>C]T 3.101973e-02 0.0190799601 0.011272241 0.016995967 0.0049901055
#> A[T>G]A 1.738741e-03 0.0012891294 0.002447997 0.001647615 0.0019429212
#> A[T>G]C 5.603887e-04 0.0008272892 0.001636789 0.001698571 0.0018057899
#> A[T>G]G 1.540888e-03 0.0026056097 0.004063937 0.006023988 0.0042585765
#> A[T>G]T 2.340882e-03 0.0021415944 0.003399764 0.005635156 0.0032752150
#> C[T>G]A 1.299297e-03 0.0005943313 0.001753389 0.001478755 0.0006888606
#> C[T>G]C 3.882152e-03 0.0030504770 0.003076258 0.006460693 0.0026525892
#> C[T>G]G 4.278988e-03 0.0035171361 0.005262728 0.007513885 0.0044746952
#> C[T>G]T 2.768471e-03 0.0013521020 0.005078980 0.025166251 0.0036828507
#> G[T>G]A 3.134337e-04 0.0006051913 0.001501838 0.002323458 0.0008937109
#> G[T>G]C 1.581823e-05 0.0005654915 0.001052993 0.003423481 0.0003260169
#> G[T>G]G 1.283198e-03 0.0036426955 0.008106156 0.010937239 0.0030755801
#> G[T>G]T 1.938235e-03 0.0019914858 0.001949606 0.009740813 0.0012274704
#> T[T>G]A 2.239748e-03 0.0022685426 0.004545048 0.003739540 0.0027913751
#> T[T>G]C 2.753271e-03 0.0023656739 0.004788050 0.004909587 0.0030796233
#> T[T>G]G 1.702656e-03 0.0022183143 0.005969082 0.007596053 0.0034624082
#> T[T>G]T 2.839907e-03 0.0027864029 0.011268331 0.011579332 0.0103274820
#> Sig6 Sig7 Sig8 Sig9 Sig10
#> A[C>A]A 3.826092e-02 4.724913e-02 0.0178120539 0.0134224026 0.0120391543
#> A[C>A]C 3.836599e-02 8.241573e-03 0.0139272839 0.0033603340 0.0065572390
#> A[C>A]G 2.094793e-03 1.877803e-03 0.0012102579 0.0007943677 0.0015375507
#> A[C>A]T 3.641442e-02 1.588556e-02 0.0153677237 0.0065087852 0.0066336276
#> C[C>A]A 2.691809e-02 8.740133e-02 0.0131271236 0.0239850321 0.0155614286
#> C[C>A]C 2.733726e-02 1.811442e-02 0.0141389819 0.0052437058 0.0188099457
#> C[C>A]G 1.666300e-04 1.669183e-02 0.0024383188 0.0049664365 0.0068906550
#> C[C>A]T 2.765704e-02 3.358228e-02 0.0206655765 0.0087933355 0.0475854887
#> G[C>A]A 1.949879e-02 1.262603e-01 0.0070080108 0.0378816538 0.0127253347
#> G[C>A]C 1.538800e-02 1.438300e-02 0.0097578021 0.0053342788 0.0082831210
#> G[C>A]G 6.004324e-04 8.823974e-03 0.0017669051 0.0033750579 0.0028465644
#> G[C>A]T 1.999634e-02 6.981223e-02 0.0108119313 0.0216895840 0.0116465007
#> T[C>A]A 2.724107e-02 6.623871e-02 0.0064147522 0.0293409653 0.0129661113
#> T[C>A]C 1.582204e-02 4.164323e-02 0.0094472583 0.0153243751 0.0100660933
#> T[C>A]G 3.532825e-04 1.443802e-02 0.0021500240 0.0056357803 0.0014633689
#> T[C>A]T 3.517898e-02 1.157670e-01 0.0168354080 0.0376292317 0.0200493195
#> A[C>G]A 9.645533e-03 1.031817e-03 0.0146673491 0.0014754693 0.0080881830
#> A[C>G]C 5.135836e-03 2.209190e-03 0.0078484306 0.0022226966 0.0042084412
#> A[C>G]G 1.451011e-10 2.597836e-03 0.0027287740 0.0008169172 0.0019582682
#> A[C>G]T 7.897040e-03 2.668708e-03 0.0127185089 0.0032522025 0.0080256929
#> C[C>G]A 5.796642e-03 5.780703e-04 0.0086263852 0.0021906998 0.0033531303
#> C[C>G]C 5.310799e-03 2.752022e-03 0.0090260821 0.0017172840 0.0044565670
#> C[C>G]G 6.237546e-04 4.876815e-03 0.0018590308 0.0018933333 0.0012296529
#> C[C>G]T 6.274642e-03 5.001674e-03 0.0136617543 0.0040023620 0.0056625123
#> G[C>G]A 4.179727e-03 6.368407e-04 0.0059739128 0.0014569313 0.0031583787
#> G[C>G]C 3.233020e-03 1.317224e-04 0.0061827944 0.0008831625 0.0039056820
#> G[C>G]G 3.580418e-05 4.119368e-04 0.0008530248 0.0002924218 0.0003746795
#> G[C>G]T 6.478774e-03 1.438490e-03 0.0096749059 0.0017629033 0.0044325580
#> T[C>G]A 2.323334e-03 3.415215e-03 0.0060449161 0.0762079493 0.0022918252
#> T[C>G]C 1.698423e-03 7.502304e-03 0.0110975814 0.0194681430 0.0054747614
#> T[C>G]G 2.484306e-08 1.419331e-03 0.0012534141 0.0032769414 0.0008197758
#> T[C>G]T 1.846115e-03 9.024908e-03 0.0151944796 0.0888325266 0.0040744115
#> A[C>T]A 2.370786e-02 2.496054e-03 0.0176940316 0.0059672125 0.0258842538
#> A[C>T]C 6.582582e-03 1.919727e-03 0.0095584856 0.0033897338 0.0154495286
#> A[C>T]G 2.088552e-02 4.928558e-03 0.0222072201 0.0069633459 0.0321735112
#> A[C>T]T 2.253107e-02 3.322242e-03 0.0153794057 0.0042141857 0.0165521323
#> C[C>T]A 4.345598e-03 1.250457e-02 0.0168959988 0.0138731641 0.0129481390
#> C[C>T]C 3.165979e-03 5.384920e-03 0.0103419142 0.0033736149 0.0119390229
#> C[C>T]G 2.821050e-03 1.344941e-02 0.0227772235 0.0081081701 0.0440187948
#> C[C>T]T 2.145700e-02 6.152561e-03 0.0164242241 0.0070786229 0.0142299520
#> G[C>T]A 1.312104e-03 1.343438e-03 0.0220089101 0.0025389091 0.0464856158
#> G[C>T]C 2.315119e-05 1.748582e-03 0.0192937463 0.0004127408 0.0548388272
#> G[C>T]G 8.744178e-03 5.723040e-03 0.0300244012 0.0054582906 0.0714191409
#> G[C>T]T 6.928898e-03 1.529667e-03 0.0167362519 0.0005565121 0.0408701927
#> T[C>T]A 5.823772e-03 4.532300e-02 0.0442265088 0.0314452596 0.0070663450
#> T[C>T]C 2.253999e-03 1.611109e-02 0.0201997571 0.0120215042 0.0147789197
#> T[C>T]G 5.537855e-03 1.261871e-02 0.0148908088 0.0108637349 0.0157875262
#> T[C>T]T 9.974362e-03 2.076265e-02 0.0325808186 0.0110713497 0.0058688621
#> A[T>A]A 1.423292e-02 2.141479e-03 0.0074303693 0.0014631776 0.0066818314
#> A[T>A]C 1.272471e-02 6.333448e-04 0.0064094606 0.0012846173 0.0069751754
#> A[T>A]G 1.540660e-02 2.947117e-03 0.0077539962 0.0020676282 0.0058077176
#> A[T>A]T 3.217455e-02 1.523081e-04 0.0061796402 0.0026443216 0.0085698150
#> C[T>A]A 1.444728e-02 3.023481e-04 0.0062117635 0.0001486237 0.0026416320
#> C[T>A]C 2.100183e-02 2.090793e-03 0.0089687822 0.0053116300 0.0069589124
#> C[T>A]G 1.961117e-02 4.420913e-03 0.0083902941 0.0037940298 0.0075472520
#> C[T>A]T 2.846925e-02 1.744077e-03 0.0156355488 0.0162482758 0.0060410818
#> G[T>A]A 1.003547e-02 1.179375e-03 0.0045826999 0.0005598006 0.0051220725
#> G[T>A]C 8.060601e-03 1.189650e-03 0.0050306421 0.0014907148 0.0046399216
#> G[T>A]G 8.900403e-03 7.816584e-04 0.0071840138 0.0021044256 0.0040972843
#> G[T>A]T 2.037261e-02 3.045276e-03 0.0056529781 0.0034971232 0.0052962854
#> T[T>A]A 1.887312e-02 4.337576e-03 0.0061573573 0.0021505975 0.0038626313
#> T[T>A]C 1.175052e-02 1.169116e-03 0.0076919701 0.0027793934 0.0037798405
#> T[T>A]G 9.486536e-03 1.285751e-03 0.0045525439 0.0004788803 0.0042715468
#> T[T>A]T 2.992503e-02 2.695800e-03 0.0140448765 0.0022560741 0.0060524261
#> A[T>C]A 1.897613e-02 3.491014e-03 0.0116281383 0.0023708518 0.0201055777
#> A[T>C]C 9.644984e-03 1.144504e-03 0.0055507127 0.0061969947 0.0051800539
#> A[T>C]G 8.834625e-03 3.963776e-03 0.0108010017 0.0040994064 0.0232799206
#> A[T>C]T 2.660401e-02 3.479688e-03 0.0122898657 0.0036679653 0.0172913151
#> C[T>C]A 4.449973e-03 1.039728e-03 0.0061863797 0.0047714222 0.0065944432
#> C[T>C]C 7.738710e-03 3.462014e-03 0.0122715112 0.0204734083 0.0062898033
#> C[T>C]G 2.261935e-03 2.447112e-03 0.0121265701 0.0149242053 0.0228292536
#> C[T>C]T 1.011002e-02 2.729870e-03 0.0179000016 0.0509213996 0.0077815527
#> G[T>C]A 3.379184e-03 6.143812e-04 0.0045004640 0.0008852611 0.0089635145
#> G[T>C]C 6.094640e-03 2.476069e-03 0.0047404046 0.0056460031 0.0070417760
#> G[T>C]G 4.415338e-03 6.347855e-03 0.0038602710 0.0042234461 0.0099671842
#> G[T>C]T 4.797926e-03 5.018722e-03 0.0063903717 0.0055672133 0.0061755795
#> T[T>C]A 3.437703e-03 9.460714e-04 0.0049596680 0.0014353270 0.0071029725
#> T[T>C]C 5.192531e-03 6.050624e-03 0.0082631437 0.0074352610 0.0043747297
#> T[T>C]G 6.655287e-04 1.290089e-03 0.0033540732 0.0041272843 0.0073112724
#> T[T>C]T 9.884785e-03 1.394912e-03 0.0058716913 0.0048466513 0.0074672744
#> A[T>G]A 5.658475e-03 1.182289e-04 0.0014011203 0.0004073580 0.0025272667
#> A[T>G]C 8.513284e-04 1.784286e-03 0.0016842477 0.0038676952 0.0020974923
#> A[T>G]G 8.160365e-03 1.625460e-03 0.0055068951 0.0026325917 0.0032523391
#> A[T>G]T 2.151774e-03 2.087405e-03 0.0082282101 0.0158771327 0.0024145507
#> C[T>G]A 3.266457e-03 4.229457e-17 0.0015611360 0.0002631693 0.0006352990
#> C[T>G]C 1.932827e-03 1.637104e-03 0.0058766170 0.0099090014 0.0030969060
#> C[T>G]G 6.208937e-03 4.746647e-03 0.0077241759 0.0085753497 0.0068729791
#> C[T>G]T 1.801532e-03 5.301436e-03 0.0352045920 0.1309119350 0.0061794503
#> G[T>G]A 5.198928e-04 1.494047e-03 0.0023336045 0.0005736685 0.0010114515
#> G[T>G]C 1.696518e-04 3.541146e-05 0.0030584286 0.0047012463 0.0012350613
#> G[T>G]G 5.370159e-03 9.631609e-03 0.0083540934 0.0049040078 0.0041473985
#> G[T>G]T 4.921895e-03 3.192798e-03 0.0113362707 0.0304442125 0.0031258182
#> T[T>G]A 1.667187e-03 2.876156e-04 0.0033834647 0.0004168605 0.0031798650
#> T[T>G]C 2.507839e-03 2.692266e-03 0.0042889353 0.0055431476 0.0032710279
#> T[T>G]G 5.720718e-03 9.819975e-04 0.0078442218 0.0031388763 0.0033484695
#> T[T>G]T 9.261816e-03 9.452530e-04 0.0181123193 0.0275887115 0.0100161880
#> Sig11
#> A[C>A]A 0.0119643388
#> A[C>A]C 0.0113081129
#> A[C>A]G 0.0021554124
#> A[C>A]T 0.0095026398
#> C[C>A]A 0.0140018781
#> C[C>A]C 0.0112085274
#> C[C>A]G 0.0033745855
#> C[C>A]T 0.0216049082
#> G[C>A]A 0.0075259551
#> G[C>A]C 0.0086974062
#> G[C>A]G 0.0011124183
#> G[C>A]T 0.0088701419
#> T[C>A]A 0.0139543764
#> T[C>A]C 0.0131944655
#> T[C>A]G 0.0019420151
#> T[C>A]T 0.0177088881
#> A[C>G]A 0.0106766175
#> A[C>G]C 0.0056850823
#> A[C>G]G 0.0017416306
#> A[C>G]T 0.0107718592
#> C[C>G]A 0.0078516501
#> C[C>G]C 0.0056832804
#> C[C>G]G 0.0019173927
#> C[C>G]T 0.0100487174
#> G[C>G]A 0.0057315401
#> G[C>G]C 0.0031656594
#> G[C>G]G 0.0009532399
#> G[C>G]T 0.0079408169
#> T[C>G]A 0.0252838614
#> T[C>G]C 0.0100958922
#> T[C>G]G 0.0016943186
#> T[C>G]T 0.0427654345
#> A[C>T]A 0.0146486965
#> A[C>T]C 0.0077349315
#> A[C>T]G 0.0087236363
#> A[C>T]T 0.0140504467
#> C[C>T]A 0.0111932318
#> C[C>T]C 0.0059472256
#> C[C>T]G 0.0145907664
#> C[C>T]T 0.0085889933
#> G[C>T]A 0.0177498640
#> G[C>T]C 0.0163100076
#> G[C>T]G 0.0192857634
#> G[C>T]T 0.0151724714
#> T[C>T]A 0.1434999874
#> T[C>T]C 0.0329228076
#> T[C>T]G 0.0171585190
#> T[C>T]T 0.0815654393
#> A[T>A]A 0.0047901312
#> A[T>A]C 0.0065566317
#> A[T>A]G 0.0062986608
#> A[T>A]T 0.0078771656
#> C[T>A]A 0.0036529121
#> C[T>A]C 0.0074921966
#> C[T>A]G 0.0074136921
#> C[T>A]T 0.0079863388
#> G[T>A]A 0.0033487924
#> G[T>A]C 0.0029406654
#> G[T>A]G 0.0038110484
#> G[T>A]T 0.0043923215
#> T[T>A]A 0.0033251823
#> T[T>A]C 0.0062149076
#> T[T>A]G 0.0028027817
#> T[T>A]T 0.0097833133
#> A[T>C]A 0.0090339055
#> A[T>C]C 0.0079815971
#> A[T>C]G 0.0105328956
#> A[T>C]T 0.0102954236
#> C[T>C]A 0.0063542993
#> C[T>C]C 0.0079246661
#> C[T>C]G 0.0105837934
#> C[T>C]T 0.0055189942
#> G[T>C]A 0.0080367755
#> G[T>C]C 0.0046364642
#> G[T>C]G 0.0043811033
#> G[T>C]T 0.0057576598
#> T[T>C]A 0.0057357268
#> T[T>C]C 0.0081113805
#> T[T>C]G 0.0058331252
#> T[T>C]T 0.0073368347
#> A[T>G]A 0.0010862997
#> A[T>G]C 0.0010026306
#> A[T>G]G 0.0042593846
#> A[T>G]T 0.0031366896
#> C[T>G]A 0.0008762220
#> C[T>G]C 0.0029019742
#> C[T>G]G 0.0080639777
#> C[T>G]T 0.0004453111
#> G[T>G]A 0.0016244383
#> G[T>G]C 0.0022854326
#> G[T>G]G 0.0065617637
#> G[T>G]T 0.0032704409
#> T[T>G]A 0.0028479597
#> T[T>G]C 0.0042639236
#> T[T>G]G 0.0043744212
#> T[T>G]T 0.0069818933
#>
#> $Exposure
#> Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 Sample_7
#> Sig1 2892.6085 1438.31588 2144.9114 3.666354e+02 99.63291 137.893150
#> Sig2 3513.9127 1630.36643 2593.6703 5.099705e+02 35.87441 206.990635
#> Sig3 883.1682 999.82449 1620.5006 2.200093e+02 62.56996 136.248454
#> Sig4 613.7551 381.77322 1432.5917 3.561856e+01 61.76187 47.319553
#> Sig5 681.6584 996.24499 258.9395 5.827967e+00 84.75080 85.340279
#> Sig6 2736.6553 5.33738 173.6779 2.305192e-11 129.05447 8.009669
#> Sig7 772.0221 414.59774 518.4493 5.527642e+02 273.26348 326.195505
#> Sig8 1427.0286 961.67686 411.5051 1.636235e+02 212.96144 108.651649
#> Sig9 816.6163 563.91365 748.6491 2.449189e+02 44.50443 238.489037
#> Sig10 1867.2844 2427.52364 369.7361 2.104454e-11 227.89468 237.757576
#> Sig11 2619.2959 950.43175 313.4014 5.956370e+02 380.72101 64.108139
#> Sample_8 Sample_9 Sample_10 Sample_11 Sample_13 Sample_14 Sample_15
#> Sig1 89.93212 488.793291 905.4543 776.4751 348.97759 1016.18747 1460.2398
#> Sig2 31.35218 598.472660 472.9686 463.9699 479.85092 769.99088 1689.8349
#> Sig3 251.11465 861.183926 970.2283 720.3104 185.36715 355.95277 1666.7762
#> Sig4 108.58510 1235.038937 462.9401 119.2712 111.28595 196.68282 834.4780
#> Sig5 1009.90122 236.079620 436.4380 1091.2295 34.94721 145.36836 884.3407
#> Sig6 90.69113 60.886372 2410.8005 243.0323 273.73171 67.65131 310.1513
#> Sig7 440.48911 8.771324 1211.6532 130.4269 261.79152 225.22257 619.3689
#> Sig8 74.70018 1188.665460 938.4633 462.4500 75.00998 321.96583 766.1233
#> Sig9 79.56263 3007.538680 299.7510 167.8243 178.97157 160.12889 1782.2534
#> Sig10 57.18481 229.424132 464.3371 586.7409 50.61674 232.50772 572.2277
#> Sig11 158.47136 209.090714 1139.9931 494.2835 116.45055 719.36049 202.2660
#> Sample_16 Sample_18 Sample_20 Sample_21 Sample_23 Sample_25 Sample_26
#> Sig1 934.2772 160.1461 480.89236 686.2181 98.51086 209.63011 151.50822
#> Sig2 879.5054 340.8865 280.00483 1764.4183 26.83737 19.20814 334.19769
#> Sig3 755.1573 241.5888 93.86510 1550.0272 129.18451 265.97631 305.58491
#> Sig4 1081.1668 404.8437 187.53047 1562.8443 175.31600 442.56548 199.63635
#> Sig5 130.6072 284.1178 302.04813 385.5179 214.21455 1262.10468 1358.37761
#> Sig6 205.8966 1624.0754 71.94879 3375.6279 446.89891 27.27933 516.71080
#> Sig7 2249.0159 115.2477 703.74847 214.4457 1509.37124 3176.21408 18.33754
#> Sig8 230.3227 615.8694 109.10612 1382.4526 320.41983 929.48746 662.46444
#> Sig9 588.9454 1461.3680 264.78930 525.8701 764.31524 2347.32498 506.38697
#> Sig10 317.4797 178.5133 92.59464 690.8966 603.39723 714.40765 1536.45567
#> Sig11 353.5959 145.2997 10.46747 1193.6668 123.51717 767.71390 355.27864
#> Sample_27 Sample_28 Sample_29 Sample_30 Sample_1 Sample_12 Sample_17
#> Sig1 2128.2169 381.74682 441.29878 455.58574 6839.9396 7947.486 5275.766766
#> Sig2 2431.8323 147.91176 313.35221 316.96902 6459.2051 6858.115 3129.978687
#> Sig3 840.3103 286.50832 159.07111 21.13038 2722.7947 5464.118 1323.193417
#> Sig4 634.5028 206.81553 377.59857 245.44283 6487.6336 8892.051 2110.857319
#> Sig5 235.9003 186.55398 453.89135 150.34295 604.8369 1415.254 7304.314115
#> Sig6 1579.0797 194.60556 639.58753 560.70139 10107.1512 1325.049 4.146147
#> Sig7 667.4846 912.54872 66.25600 26.37378 952.4554 7312.888 559.804630
#> Sig8 408.5010 174.18293 384.03014 268.64216 3527.7995 2818.241 1671.982139
#> Sig9 604.7606 201.24096 709.88011 245.75308 1172.6965 2710.134 1397.714490
#> Sig10 257.2629 204.38831 54.05094 524.62055 1185.7381 1853.601 2977.616059
#> Sig11 302.2091 76.50792 48.95576 103.42140 2699.6698 3371.060 1916.421730
#> Sample_19 Sample_22 Sample_24
#> Sig1 5949.4895 4440.5748 5563.813
#> Sig2 3715.5190 1911.4240 7325.591
#> Sig3 3370.2751 6739.2437 7930.720
#> Sig4 2583.7245 3218.1267 4697.443
#> Sig5 4702.6021 7760.7238 2369.467
#> Sig6 1899.4088 2889.8528 2651.562
#> Sig7 5755.0451 419.3292 3705.740
#> Sig8 986.0466 4175.0721 3033.644
#> Sig9 1901.6070 727.1271 3134.564
#> Sig10 1259.3749 3983.8709 2613.616
#> Sig11 768.9564 2090.7489 2870.124
#>
#> $Exposure.norm
#> Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 Sample_7
#> Sig1 0.15366594 0.1335482891 0.20261712 1.360426e-01 0.06176910 0.086344919
#> Sig2 0.18667189 0.1513802711 0.24500872 1.892280e-01 0.02224094 0.129611874
#> Sig3 0.04691712 0.0928341628 0.15307913 8.163593e-02 0.03879130 0.085315055
#> Sig4 0.03260491 0.0354478183 0.13532848 1.321651e-02 0.03829031 0.029630210
#> Sig5 0.03621218 0.0925018045 0.02446049 2.162507e-03 0.05254269 0.053437748
#> Sig6 0.14538114 0.0004955782 0.01640633 8.553571e-15 0.08000949 0.005015436
#> Sig7 0.04101264 0.0384955900 0.04897485 2.051069e-01 0.16941430 0.204254703
#> Sig8 0.07580898 0.0892921373 0.03887246 6.071361e-02 0.13202903 0.068034691
#> Sig9 0.04338164 0.0523596412 0.07072046 9.087881e-02 0.02759127 0.149335311
#> Sig10 0.09919697 0.2253966837 0.03492679 7.808719e-15 0.14128715 0.148877291
#> Sig11 0.13914658 0.0882480240 0.02960518 2.210151e-01 0.23603441 0.040142763
#> Sample_8 Sample_9 Sample_10 Sample_11 Sample_13 Sample_14
#> Sig1 0.03759728 0.060166986 0.09322060 0.14773080 0.16484527 0.24131628
#> Sig2 0.01310718 0.073667738 0.04869426 0.08827409 0.22666543 0.18285143
#> Sig3 0.10498172 0.106005631 0.09988938 0.13704499 0.08756121 0.08452889
#> Sig4 0.04539540 0.152024530 0.04766177 0.02269233 0.05256774 0.04670670
#> Sig5 0.42220225 0.029059726 0.04493326 0.20761541 0.01650789 0.03452094
#> Sig6 0.03791460 0.007494680 0.24820278 0.04623890 0.12930165 0.01606531
#> Sig7 0.18415216 0.001079688 0.12474517 0.02481480 0.12366151 0.05348410
#> Sig8 0.03122938 0.146316284 0.09661903 0.08798492 0.03543219 0.07645794
#> Sig9 0.03326219 0.370206671 0.03086071 0.03192996 0.08454015 0.03802616
#> Sig10 0.02390685 0.028240483 0.04780560 0.11163229 0.02390965 0.05521412
#> Sig11 0.06625100 0.025737583 0.11736744 0.09404151 0.05500732 0.17082812
#> Sample_15 Sample_16 Sample_18 Sample_20 Sample_21 Sample_23
#> Sig1 0.13535703 0.12092684 0.02874144 0.185172566 0.05147156 0.022328024
#> Sig2 0.15663936 0.11383754 0.06117896 0.107818750 0.13234475 0.006082837
#> Sig3 0.15450194 0.09774272 0.04335799 0.036143725 0.11626379 0.029280375
#> Sig4 0.07735200 0.13993929 0.07265737 0.072210544 0.11722517 0.039736328
#> Sig5 0.08197403 0.01690496 0.05099067 0.116306752 0.02891677 0.048552896
#> Sig6 0.02874950 0.02664993 0.29147310 0.027704626 0.25319769 0.101292076
#> Sig7 0.05741244 0.29109818 0.02068352 0.270985615 0.01608505 0.342107228
#> Sig8 0.07101585 0.02981150 0.11053019 0.042012437 0.10369443 0.072624903
#> Sig9 0.16520610 0.07622931 0.26227197 0.101959855 0.03944424 0.173236219
#> Sig10 0.05304269 0.04109254 0.03203781 0.035654522 0.05182248 0.136763274
#> Sig11 0.01874906 0.04576719 0.02607697 0.004030609 0.08953406 0.027995840
#> Sample_25 Sample_26 Sample_27 Sample_28 Sample_29 Sample_30
#> Sig1 0.020629002 0.025485244 0.21092211 0.12840412 0.12097097 0.156076860
#> Sig2 0.001890210 0.056215497 0.24101266 0.04975151 0.08589763 0.108588843
#> Sig3 0.026173844 0.051402532 0.08328100 0.09636975 0.04360535 0.007238951
#> Sig4 0.043551398 0.033580893 0.06288394 0.06956434 0.10350916 0.084085041
#> Sig5 0.124199527 0.228493118 0.02337948 0.06274918 0.12442291 0.051505245
#> Sig6 0.002684468 0.086916083 0.15649853 0.06545740 0.17532685 0.192087908
#> Sig7 0.312560672 0.003084564 0.06615268 0.30694430 0.01816242 0.009035262
#> Sig8 0.091467772 0.111433348 0.04048548 0.05858806 0.10527221 0.092032784
#> Sig9 0.230992450 0.085179509 0.05993627 0.06768928 0.19459580 0.084191330
#> Sig10 0.070302483 0.258447683 0.02549667 0.06874792 0.01481671 0.179727151
#> Sig11 0.075548174 0.059761530 0.02995117 0.02573415 0.01341999 0.035430625
#> Sample_1 Sample_12 Sample_17 Sample_19 Sample_22 Sample_24
#> Sig1 0.15996147 0.15905152 0.190655021 0.18087926 0.11577234 0.12122579
#> Sig2 0.15105746 0.13725016 0.113110791 0.11296101 0.04983365 0.15961185
#> Sig3 0.06367633 0.10935234 0.047817404 0.10246473 0.17570203 0.17279656
#> Sig4 0.15172230 0.17795493 0.076281907 0.07855164 0.08390131 0.10234909
#> Sig5 0.01414495 0.02832321 0.263962420 0.14297078 0.20233353 0.05162655
#> Sig6 0.23636974 0.02651795 0.000149833 0.05774675 0.07534273 0.05777290
#> Sig7 0.02227449 0.14635143 0.020230152 0.17496767 0.01093253 0.08074162
#> Sig8 0.08250248 0.05640091 0.060421888 0.02997827 0.10885029 0.06609781
#> Sig9 0.02742513 0.05423740 0.050510437 0.05781358 0.01895728 0.06829669
#> Sig10 0.02773013 0.03709576 0.107604729 0.03828813 0.10386540 0.05694613
#> Sig11 0.06313552 0.06746439 0.069255417 0.02337818 0.05450891 0.06253499
#>
#> $K
#> [1] 11
#>
#> attr(,"class")
#> [1] "Signature"
#> attr(,"used_runs")
#> [1] 6
#> attr(,"method")
#> [1] "brunet"
#> attr(,"call_method")
#> [1] "NMF with best practice"
#> attr(,"nrun")
#> [1] 50
#> attr(,"seed")
#> [1] 123456
# If you think the suggested signature number is not right
# Just pick up the solution you want
obj_s8 <- bp_get_sig_obj(e1, 8)
# Track the reconstructed profile similarity
rec_sim <- get_sig_rec_similarity(obj_s8, t(simulated_catalogs$set1))
rec_sim
#> sample similarity rss unexplained_variance
#> <char> <num> <num> <num>
#> 1: Sample_1 0.9997486 13230.30099 0.0005076483
#> 2: Sample_2 0.9992611 20030.77983 0.0014806239
#> 3: Sample_3 0.9903052 59263.95137 0.0196886410
#> 4: Sample_4 0.9997825 1856.76699 0.0004676679
#> 5: Sample_5 0.9992557 1019.54200 0.0015201050
#> 6: Sample_6 0.9958607 721.60675 0.0083609296
#> 7: Sample_7 0.9982163 261.63888 0.0036855218
#> 8: Sample_8 0.9801222 7122.56156 0.0410276351
#> 9: Sample_9 0.9992708 3331.53459 0.0014817779
#> 10: Sample_10 0.9989004 3735.70983 0.0022051099
#> 11: Sample_11 0.9993768 885.02187 0.0012459902
#> 12: Sample_12 0.9990641 77738.85426 0.0019042753
#> 13: Sample_13 0.9997332 87.78296 0.0005511132
#> 14: Sample_14 0.9995498 805.36015 0.0010261993
#> 15: Sample_15 0.9991823 5885.65491 0.0016497945
#> 16: Sample_16 0.9994722 1343.32088 0.0010669067
#> 17: Sample_17 0.9994158 24812.49173 0.0012580570
#> 18: Sample_18 0.9969664 4761.54340 0.0060580974
#> 19: Sample_19 0.9979704 77097.52442 0.0040672214
#> 20: Sample_20 0.9984978 397.72548 0.0030309129
#> 21: Sample_21 0.9995502 3540.17917 0.0009081402
#> 22: Sample_22 0.9996566 15566.63589 0.0006927896
#> 23: Sample_23 0.9973757 2453.12111 0.0052897034
#> 24: Sample_24 0.9999582 4645.77103 0.0000843955
#> 25: Sample_25 0.9996526 2058.53068 0.0006979333
#> 26: Sample_26 0.9950134 10587.56050 0.0100460865
#> 27: Sample_27 0.9992121 4574.39411 0.0015861921
#> 28: Sample_28 0.9983929 576.01938 0.0032494451
#> 29: Sample_29 0.9966799 1831.31566 0.0066974197
#> 30: Sample_30 0.9913698 2563.94388 0.0174160856
#> sample similarity rss unexplained_variance
#> <char> <num> <num> <num>
# After extraction, you can assign the signatures
# to reference COSMIC signatures
# More see ?get_sig_similarity
sim <- get_sig_similarity(obj_suggested)
#> -Comparing against COSMIC signatures
#> ------------------------------------
#> --Found Sig1 most similar to SBS12
#> Aetiology: Unknown [similarity: 0.827]
#> --Found Sig2 most similar to SBS13
#> Aetiology: Activity of APOBEC family of cytidine deaminases [similarity: 0.881]
#> --Found Sig3 most similar to SBS40
#> Aetiology: Unknown [similarity: 0.768]
#> --Found Sig4 most similar to SBS3
#> Aetiology: Defective homologous recombination DNA damage repair [similarity: 0.901]
#> --Found Sig5 most similar to SBS1
#> Aetiology: Spontaneous deamination of 5-methylcytosine (clock-like signature) [similarity: 0.917]
#> --Found Sig6 most similar to SBS8
#> Aetiology: Unknown [similarity: 0.939]
#> --Found Sig7 most similar to SBS18
#> Aetiology: Damage by reactive oxygen species [similarity: 0.975]
#> --Found Sig8 most similar to SBS40
#> Aetiology: Unknown [similarity: 0.849]
#> --Found Sig9 most similar to SBS17b
#> Aetiology: Unknown [similarity: 0.67]
#> --Found Sig10 most similar to SBS6
#> Aetiology: Defective DNA mismatch repair [similarity: 0.828]
#> --Found Sig11 most similar to SBS2
#> Aetiology: Activity of APOBEC family of cytidine deaminases [similarity: 0.872]
#> ------------------------------------
#> Return result invisiblely.
# Visualize the match result
if (require(pheatmap)) {
pheatmap::pheatmap(sim$similarity)
}
#> Loading required package: pheatmap
# Obtain the suggested solution from extraction result
obj_suggested <- bp_get_sig_obj(e1, e1$suggested)
obj_suggested
#> $Signature
#> Sig1 Sig2 Sig3 Sig4 Sig5 Sig6
#> A[C>A]A 364.6855115 346.94762 452.99236 692.34882 380.33725 1.324947e+03
#> A[C>A]C 144.9048999 252.82263 388.79723 440.77833 227.80360 1.328586e+03
#> A[C>A]G 101.9965735 56.31129 73.55796 77.30450 35.81481 7.254115e+01
#> A[C>A]T 228.2381304 215.37118 321.71075 477.84538 175.07128 1.261005e+03
#> C[C>A]A 453.0228668 373.09497 354.26747 877.39212 132.35013 9.321536e+02
#> C[C>A]C 352.2870235 356.88211 261.08852 585.38882 103.33427 9.466691e+02
#> C[C>A]G 139.5844540 77.21084 71.37286 89.08911 67.52359 5.770273e+00
#> C[C>A]T 206.6452684 325.06644 233.99811 449.18696 224.99675 9.577429e+02
#> G[C>A]A 262.9030436 93.42677 249.76023 671.83989 292.79971 6.752285e+02
#> G[C>A]C 234.4691391 203.94031 338.46614 341.08226 239.81114 5.328750e+02
#> G[C>A]G 77.2748163 38.76242 67.73828 55.54570 33.87816 2.079253e+01
#> G[C>A]T 25.1498022 147.95376 306.35190 471.88666 250.35555 6.924583e+02
#> T[C>A]A 707.9694364 1128.97194 915.45972 578.90913 425.73686 9.433380e+02
#> T[C>A]C 550.3665329 639.22968 560.35636 691.10963 363.64965 5.479057e+02
#> T[C>A]G 124.1977001 90.18331 156.15108 117.38406 93.80205 1.223391e+01
#> T[C>A]T 423.1960918 487.78631 622.24017 648.89415 477.28252 1.218222e+03
#> A[C>G]A 191.6864937 322.45523 599.97352 496.63443 220.62923 3.340177e+02
#> A[C>G]C 98.4509868 163.72693 305.70556 240.58008 192.32289 1.778502e+02
#> A[C>G]G 32.9514639 41.90439 110.29973 130.08351 51.99749 5.024744e-06
#> A[C>G]T 196.5392614 354.14978 543.65236 573.08529 259.68447 2.734687e+02
#> C[C>G]A 170.7588970 286.52200 405.42625 425.08854 151.77176 2.007334e+02
#> C[C>G]C 184.4906424 173.75603 306.77347 324.29104 135.08277 1.839090e+02
#> C[C>G]G 37.1196263 34.60268 108.73587 98.87709 47.68770 2.160016e+01
#> C[C>G]T 191.1959730 337.55583 526.49460 653.67282 177.24473 2.172862e+02
#> G[C>G]A 120.8305851 147.81819 245.41235 278.24330 94.40144 1.447409e+02
#> G[C>G]C 85.9454946 129.07501 248.42925 244.06828 203.54229 1.119571e+02
#> G[C>G]G 11.2368880 18.09011 48.93059 39.78653 18.81565 1.239872e+00
#> G[C>G]T 99.2539804 194.17868 318.01486 404.69066 119.83849 2.243552e+02
#> T[C>G]A 2304.8190218 5806.53390 2543.20013 827.06428 4.47700 8.045533e+01
#> T[C>G]C 649.4320013 1498.46533 908.51263 607.96705 148.26065 5.881513e+01
#> T[C>G]G 91.1666377 209.73580 147.99082 71.38912 23.82618 8.602967e-04
#> T[C>G]T 2761.4204889 6844.53103 3311.67302 1324.25369 108.29443 6.392960e+01
#> A[C>T]A 580.6367660 486.55883 771.14239 573.23727 1045.30267 8.209856e+02
#> A[C>T]C 336.8462426 166.00633 384.82160 259.72339 579.17288 2.279499e+02
#> A[C>T]G 41.2744554 48.63027 1095.73657 279.15822 5056.19068 7.232503e+02
#> A[C>T]T 365.5627960 320.98049 532.98918 433.35276 478.77227 7.802344e+02
#> C[C>T]A 1107.9997769 578.99821 714.60791 489.10670 828.90566 1.504848e+02
#> C[C>T]C 680.5296357 284.55116 583.37633 291.95102 705.17673 1.096355e+02
#> C[C>T]G 140.6997922 110.49878 608.69038 382.82184 2856.59934 9.769086e+01
#> C[C>T]T 899.7884560 535.31877 570.95299 596.89330 769.16400 7.430400e+02
#> G[C>T]A 740.5029427 445.50554 653.16343 432.39515 1022.83916 4.543718e+01
#> G[C>T]C 518.2147424 336.90086 473.33371 288.04137 1073.90995 8.017087e-01
#> G[C>T]G 151.0536663 97.06964 535.59679 111.37791 3150.67238 3.028044e+02
#> G[C>T]T 684.1239448 427.63207 522.89528 260.30904 744.80860 2.399426e+02
#> T[C>T]A 1935.5135649 2371.00890 1254.10598 184.58238 468.56327 2.016729e+02
#> T[C>T]C 808.7068291 534.84839 784.12753 396.90968 736.96715 7.805433e+01
#> T[C>T]G 174.8872796 75.86828 502.79237 130.24016 2179.16906 1.917718e+02
#> T[C>T]T 887.6696088 710.46176 664.34758 223.84417 452.25297 3.454048e+02
#> A[T>A]A 317.6552554 228.37936 246.26228 271.09229 168.31318 4.928755e+02
#> A[T>A]C 278.2529058 213.13425 260.76926 232.40386 98.31839 4.406474e+02
#> A[T>A]G 249.1917651 182.23415 340.62694 225.47731 185.96380 5.335190e+02
#> A[T>A]T 246.2567101 310.95073 335.99184 190.22561 288.31773 1.114181e+03
#> C[T>A]A 100.2452810 96.44490 181.15835 160.06344 66.32999 5.002986e+02
#> C[T>A]C 315.3312658 290.19171 210.47003 393.73526 34.30914 7.272778e+02
#> C[T>A]G 214.4139116 134.29338 313.33861 255.20058 152.95544 6.791202e+02
#> C[T>A]T 255.1055329 189.25835 347.36489 510.57007 130.67930 9.858692e+02
#> G[T>A]A 78.5708695 46.54815 181.40288 119.84166 92.06006 3.475210e+02
#> G[T>A]C 162.1735392 152.29781 122.00953 162.13346 53.23410 2.791327e+02
#> G[T>A]G 159.3889064 110.89872 247.32946 192.69463 94.97434 3.082144e+02
#> G[T>A]T 115.6398251 140.47073 153.38354 297.73054 26.48535 7.054885e+02
#> T[T>A]A 252.4670358 160.68865 305.17529 216.26710 228.23242 6.535623e+02
#> T[T>A]C 592.0449289 353.72488 228.79198 337.76826 55.16387 4.069120e+02
#> T[T>A]G 198.0755104 151.91335 256.84425 226.22452 62.18204 3.285117e+02
#> T[T>A]T 383.5737823 337.00589 351.72460 404.10447 88.98538 1.036281e+03
#> A[T>C]A 2214.2964522 1259.06003 851.26760 769.08597 645.92386 6.571294e+02
#> A[T>C]C 1296.4130889 789.71635 429.95798 612.10931 168.38083 3.339987e+02
#> A[T>C]G 2184.7595181 1231.70050 712.42662 866.79096 423.38503 3.059365e+02
#> A[T>C]T 1334.2884205 746.77707 823.13917 536.52760 381.41889 9.212770e+02
#> C[T>C]A 1368.4259217 768.78881 352.44770 550.53435 178.86216 1.540993e+02
#> C[T>C]C 1517.2716800 984.20451 361.66402 904.29318 97.95732 2.679858e+02
#> C[T>C]G 1890.4159043 1020.51180 435.96306 693.79180 283.74277 7.832913e+01
#> C[T>C]T 1419.4825104 789.41692 461.84585 894.63908 253.60587 3.501025e+02
#> G[T>C]A 2449.4241104 1446.44266 424.35425 906.38195 218.56288 1.170186e+02
#> G[T>C]C 1114.6078783 664.36882 219.30814 553.12788 124.96875 2.110529e+02
#> G[T>C]G 1737.5192128 1021.54590 349.01345 654.90212 130.34669 1.528999e+02
#> G[T>C]T 1758.5622876 1017.16814 477.82218 719.38885 245.19309 1.661486e+02
#> T[T>C]A 2185.6729708 1226.57228 480.33544 787.78432 208.84153 1.190451e+02
#> T[T>C]C 1490.7693030 863.31094 418.77913 676.08017 163.41359 1.798135e+02
#> T[T>C]G 1357.3556066 889.25467 370.53252 521.65067 232.69739 2.304677e+01
#> T[T>C]T 1684.7172024 939.72966 463.64985 665.37852 175.01269 3.423028e+02
#> A[T>G]A 94.4330523 63.49243 100.69104 64.50282 68.14202 1.959488e+02
#> A[T>G]C 30.4353615 40.74580 67.32443 66.49769 63.33256 2.948087e+01
#> A[T>G]G 83.6873843 128.33196 167.15785 235.83434 149.35654 2.825874e+02
#> A[T>G]T 127.1360117 105.47819 139.83909 220.61187 114.86815 7.451436e+01
#> C[T>G]A 70.5662971 29.27212 72.12039 57.89206 24.15968 1.131150e+02
#> C[T>G]C 210.8441666 150.24265 126.53264 252.93096 93.03145 6.693238e+01
#> C[T>G]G 232.3968048 173.22663 216.46656 294.16258 156.93625 2.150109e+02
#> C[T>G]T 150.3588432 66.59398 208.90862 985.23861 129.16472 6.238572e+01
#> G[T>G]A 17.0229495 29.80699 61.77359 90.96154 31.34418 1.800350e+01
#> G[T>G]C 0.8591066 27.85169 43.31172 134.02653 11.43404 5.874917e+00
#> G[T>G]G 69.6919626 179.41070 333.42242 428.18415 107.86656 1.859646e+02
#> G[T>G]T 105.2678056 98.08502 80.19119 381.34504 43.04977 1.704416e+02
#> T[T>G]A 121.6433012 111.73067 186.94694 146.40000 97.89894 5.773345e+01
#> T[T>G]C 149.5333411 116.51460 196.94210 192.20642 108.00836 8.684462e+01
#> T[T>G]G 92.4731954 109.25682 245.52028 297.37939 121.43337 1.981042e+02
#> T[T>G]T 154.2386196 137.23642 463.48903 453.32162 362.20483 3.207298e+02
#> Sig7 Sig8 Sig9 Sig10 Sig11
#> A[C>A]A 1.612108e+03 513.18466 370.426103 317.485977 301.03623
#> A[C>A]C 2.811969e+02 401.26021 92.737155 172.921734 284.52485
#> A[C>A]G 6.406936e+01 34.86885 21.922641 40.546934 54.23260
#> A[C>A]T 5.420044e+02 442.76085 179.626853 174.936185 239.09711
#> C[C>A]A 2.982074e+03 378.20672 661.929329 410.372295 352.30301
#> C[C>A]C 6.180518e+02 407.35946 144.713698 496.039327 282.01916
#> C[C>A]G 5.695139e+02 70.25062 137.061730 181.714287 84.90837
#> C[C>A]T 1.145804e+03 595.39775 242.674959 1254.882610 543.60381
#> G[C>A]A 4.307915e+03 201.90842 1045.442740 335.581323 189.36150
#> G[C>A]C 4.907381e+02 281.13290 147.213294 218.435173 218.83653
#> G[C>A]G 3.010680e+02 50.90646 93.143500 75.067089 27.98970
#> G[C>A]T 2.381946e+03 311.50351 598.580470 307.131262 223.18275
#> T[C>A]A 2.260020e+03 184.81599 809.740232 341.930871 351.10781
#> T[C>A]C 1.420839e+03 272.18579 422.915978 265.454149 331.98760
#> T[C>A]G 4.926154e+02 61.94453 155.534013 38.590677 48.86329
#> T[C>A]T 3.949891e+03 485.04643 1038.476495 528.723002 445.57556
#> A[C>G]A 3.520491e+01 422.58229 40.719413 213.294440 268.63571
#> A[C>G]C 7.537606e+01 226.12183 61.341091 110.981307 143.04307
#> A[C>G]G 8.863639e+01 78.61895 22.544953 51.641726 43.82139
#> A[C>G]T 9.105450e+01 366.43408 89.752984 211.646507 271.03210
#> C[C>G]A 1.972337e+01 248.53555 60.458058 88.425799 197.55635
#> C[C>G]C 9.389712e+01 260.05125 47.392917 117.524661 142.99773
#> C[C>G]G 1.663936e+02 53.56070 52.251455 32.427322 48.24376
#> C[C>G]T 1.706537e+02 393.61001 110.455586 149.326789 252.83704
#> G[C>G]A 2.172857e+01 172.11493 40.207808 83.289982 144.21200
#> G[C>G]C 4.494280e+00 178.13304 24.373164 102.997208 79.65155
#> G[C>G]G 1.405500e+01 24.57657 8.070140 9.880719 23.98459
#> G[C>G]T 4.908029e+01 278.74457 48.651901 116.891518 199.79989
#> T[C>G]A 1.165248e+02 174.16061 2103.156522 60.437995 636.17041
#> T[C>G]C 2.559735e+02 319.73340 537.274027 144.375587 254.02401
#> T[C>G]G 4.842660e+01 36.11222 90.435720 21.618406 42.63096
#> T[C>G]T 3.079238e+02 437.76949 2451.564561 107.446794 1076.02647
#> A[C>T]A 8.516367e+01 509.78431 164.680745 682.596751 368.57769
#> A[C>T]C 6.549977e+01 275.39037 93.548518 407.421365 194.61958
#> A[C>T]G 1.681590e+02 639.81418 192.171636 848.451511 219.49651
#> A[C>T]T 1.133526e+02 443.09742 116.301412 436.498259 353.52505
#> C[C>T]A 4.266475e+02 486.79212 382.866038 341.456923 281.63431
#> C[C>T]C 1.837298e+02 297.96180 93.103677 314.845399 149.63889
#> C[C>T]G 4.588847e+02 656.23660 223.766036 1160.824899 367.12010
#> C[C>T]T 2.099212e+02 473.19977 195.353006 375.259764 216.10874
#> G[C>T]A 4.583719e+01 634.10065 70.067798 1225.877730 446.60656
#> G[C>T]C 5.966043e+01 555.87383 11.390655 1446.161268 410.37815
#> G[C>T]G 1.952663e+02 865.03568 150.635721 1883.402702 485.25151
#> G[C>T]T 5.219121e+01 482.18963 15.358399 1077.792737 381.75646
#> T[C>T]A 1.546390e+03 1274.21386 867.813705 186.347428 3610.62119
#> T[C>T]C 5.496995e+02 581.97699 331.764668 389.736658 828.37489
#> T[C>T]G 4.305419e+02 429.02041 299.813014 416.334741 431.72765
#> T[C>T]T 7.084075e+02 938.68885 305.542684 154.768464 2052.27825
#> A[T>A]A 7.306582e+01 214.07703 40.380191 176.207375 120.52509
#> A[T>A]C 2.160929e+01 184.66354 35.452354 183.943184 164.97223
#> A[T>A]G 1.005536e+02 223.40107 57.061578 153.156016 158.48139
#> A[T>A]T 5.196649e+00 178.04216 72.976932 225.995616 198.19835
#> C[T>A]A 1.031591e+01 178.96767 4.101657 69.662794 91.91138
#> C[T>A]C 7.133643e+01 258.40038 146.588241 183.514312 188.51210
#> C[T>A]G 1.508386e+02 241.73351 104.706117 199.029485 186.53684
#> C[T>A]T 5.950672e+01 450.47718 448.413420 159.310090 200.94527
#> G[T>A]A 4.023947e+01 132.03257 15.449153 135.074786 84.25939
#> G[T>A]C 4.059007e+01 144.93827 41.140153 122.359928 73.99045
#> G[T>A]G 2.666966e+01 206.97926 58.077096 108.049975 95.89027
#> G[T>A]T 1.039028e+02 162.86845 96.512208 139.668975 110.51575
#> T[T>A]A 1.479951e+02 177.40016 59.351331 101.861911 83.66533
#> T[T>A]C 3.988944e+01 221.61403 76.704589 99.678627 156.37407
#> T[T>A]G 4.386895e+01 131.16375 13.215947 112.645471 70.52114
#> T[T>A]T 9.197887e+01 404.64818 62.262232 159.609253 246.15917
#> A[T>C]A 1.191110e+02 335.01932 65.429821 530.206594 227.30323
#> A[T>C]C 3.904971e+01 159.92207 171.022183 136.603821 200.82597
#> A[T>C]G 1.352414e+02 311.18862 113.133780 613.917569 265.01951
#> A[T>C]T 1.187246e+02 354.08441 101.227039 455.991338 259.04444
#> C[T>C]A 3.547481e+01 178.23633 131.679803 173.902849 159.88132
#> C[T>C]C 1.181216e+02 353.55559 565.016939 165.869156 199.39352
#> C[T>C]G 8.349381e+01 349.37968 411.872251 602.032977 266.30015
#> C[T>C]T 9.314132e+01 515.71853 1405.308433 205.208256 138.86411
#> G[T>C]A 2.096227e+01 129.66327 24.431083 236.377912 202.21432
#> G[T>C]C 8.448178e+01 136.57622 155.816136 185.699517 116.65866
#> G[T>C]G 2.165845e+02 111.21861 116.556976 262.845807 110.23349
#> G[T>C]T 1.712354e+02 184.11356 153.641728 162.856944 144.86920
#> T[T>C]A 3.227932e+01 142.89343 39.611581 187.313337 144.31734
#> T[T>C]C 2.064432e+02 238.07017 205.195362 115.366520 204.09146
#> T[T>C]G 4.401696e+01 96.63450 113.903142 192.806438 146.76799
#> T[T>C]T 4.759346e+01 169.16982 133.755945 196.920385 184.60302
#> A[T>G]A 4.033890e+00 40.36780 11.242104 66.646853 27.33252
#> A[T>G]C 6.087864e+01 48.52501 106.739107 55.313219 25.22731
#> A[T>G]G 5.545958e+01 158.65964 72.653213 85.767824 107.17091
#> A[T>G]T 7.122083e+01 237.06369 438.170763 63.674406 78.92264
#> C[T>G]A 1.443062e-12 44.97803 7.262841 16.753545 22.04673
#> C[T>G]C 5.585688e+01 169.31173 273.464660 81.668878 73.01694
#> C[T>G]G 1.619524e+02 222.54191 236.659074 181.248153 202.89875
#> C[T>G]T 1.808814e+02 1014.28261 3612.855255 162.959024 11.20453
#> G[T>G]A 5.097588e+01 67.23368 15.831875 26.673109 40.87270
#> G[T>G]C 1.208215e+00 88.11666 129.743115 32.569949 57.50406
#> G[T>G]G 3.286240e+02 240.69052 135.338848 109.371542 165.10136
#> G[T>G]T 1.089361e+02 326.61030 840.187207 82.431326 82.28797
#> T[T>G]A 9.813250e+00 97.48130 11.504350 83.856599 71.65787
#> T[T>G]C 9.185830e+01 123.56889 152.977572 86.260669 107.28512
#> T[T>G]G 3.350509e+01 226.00057 86.625452 88.302890 110.06536
#> T[T>G]T 3.225139e+01 521.83563 761.382232 264.138090 175.67229
#>
#> $Signature.norm
#> Sig1 Sig2 Sig3 Sig4 Sig5
#> A[C>A]A 6.714744e-03 0.0070443097 0.011013137 0.017684877 0.0108444883
#> A[C>A]C 2.668050e-03 0.0051332270 0.009452426 0.011258936 0.0064953234
#> A[C>A]G 1.878004e-03 0.0011433259 0.001788339 0.001974612 0.0010211813
#> A[C>A]T 4.202417e-03 0.0043728252 0.007821422 0.012205750 0.0049917763
#> C[C>A]A 8.341249e-03 0.0075751968 0.008612940 0.022411495 0.0037736757
#> C[C>A]C 6.486458e-03 0.0072460163 0.006347576 0.014952765 0.0029463516
#> C[C>A]G 2.570088e-03 0.0015676634 0.001735215 0.002275630 0.0019252882
#> C[C>A]T 3.804840e-03 0.0066000415 0.005688955 0.011473720 0.0064152924
#> G[C>A]A 4.840682e-03 0.0018969062 0.006072163 0.017161012 0.0083485461
#> G[C>A]C 4.317145e-03 0.0041407367 0.008228778 0.008712368 0.0068376923
#> G[C>A]G 1.422817e-03 0.0007870193 0.001646851 0.001418821 0.0009659619
#> G[C>A]T 4.630688e-04 0.0030040043 0.007448018 0.012053545 0.0071383433
#> T[C>A]A 1.303543e-02 0.0229222728 0.022256629 0.014787253 0.0121389593
#> T[C>A]C 1.013358e-02 0.0129787080 0.013623367 0.017653224 0.0103686778
#> T[C>A]G 2.286781e-03 0.0018310520 0.003796340 0.002998377 0.0026745613
#> T[C>A]T 7.792065e-03 0.0099038519 0.015127884 0.016574901 0.0136086717
#> A[C>G]A 3.529413e-03 0.0065470243 0.014586538 0.012685685 0.0062907618
#> A[C>G]C 1.812721e-03 0.0033242574 0.007432304 0.006145210 0.0054836684
#> A[C>G]G 6.067163e-04 0.0008508129 0.002681604 0.003322763 0.0014825952
#> A[C>G]T 3.618764e-03 0.0071905401 0.013217260 0.014638493 0.0074043371
#> C[C>G]A 3.144085e-03 0.0058174479 0.009856711 0.010858166 0.0043274412
#> C[C>G]C 3.396920e-03 0.0035278849 0.007458267 0.008283465 0.0038515909
#> C[C>G]G 6.834623e-04 0.0007025615 0.002643583 0.002525648 0.0013597108
#> C[C>G]T 3.520381e-03 0.0068536218 0.012800121 0.016696964 0.0050537474
#> G[C>G]A 2.224784e-03 0.0030012516 0.005966458 0.007107254 0.0026916513
#> G[C>G]C 1.582465e-03 0.0026206963 0.006039804 0.006234311 0.0058035652
#> G[C>G]G 2.068983e-04 0.0003672956 0.001189599 0.001016280 0.0005364872
#> G[C>G]T 1.827506e-03 0.0039425397 0.007731568 0.010337137 0.0034169337
#> T[C>G]A 4.243730e-02 0.1178939434 0.061830204 0.021125956 0.0001276519
#> T[C>G]C 1.195762e-02 0.0304243444 0.022087731 0.015529488 0.0042273295
#> T[C>G]G 1.678599e-03 0.0042584064 0.003597948 0.001823514 0.0006793516
#> T[C>G]T 5.084444e-02 0.1389690939 0.080513294 0.033825817 0.0030877799
#> A[C>T]A 1.069093e-02 0.0098789295 0.018747990 0.014642375 0.0298045291
#> A[C>T]C 6.202155e-03 0.0033705377 0.009355771 0.006634194 0.0165138533
#> A[C>T]G 7.599627e-04 0.0009873728 0.026639514 0.007130624 0.1441662650
#> A[C>T]T 6.730897e-03 0.0065170816 0.012958017 0.011069262 0.0136511486
#> C[C>T]A 2.040096e-02 0.0117557881 0.017373526 0.012493402 0.0236344396
#> C[C>T]C 1.253020e-02 0.0057774325 0.014183028 0.007457394 0.0201065787
#> C[C>T]G 2.590624e-03 0.0022435307 0.014798462 0.009778535 0.0814497087
#> C[C>T]T 1.656729e-02 0.0108689351 0.013880992 0.015246628 0.0219310362
#> G[C>T]A 1.363445e-02 0.0090453971 0.015879689 0.011044801 0.0291640310
#> G[C>T]C 9.541589e-03 0.0068403236 0.011507675 0.007357529 0.0306202032
#> G[C>T]G 2.781264e-03 0.0019708699 0.013021413 0.002844960 0.0898345608
#> G[C>T]T 1.259638e-02 0.0086825002 0.012712614 0.006649153 0.0212365951
#> T[C>T]A 3.563749e-02 0.0481401803 0.030489786 0.004714844 0.0133600610
#> T[C>T]C 1.489025e-02 0.0108593848 0.019063685 0.010138385 0.0210130133
#> T[C>T]G 3.220099e-03 0.0015404044 0.012223873 0.003326764 0.0621342595
#> T[C>T]T 1.634415e-02 0.0144249805 0.016151598 0.005717720 0.0128950083
#> A[T>A]A 5.848803e-03 0.0046369389 0.005987121 0.006924593 0.0047990836
#> A[T>A]C 5.123310e-03 0.0043274074 0.006339814 0.005936363 0.0028033349
#> A[T>A]G 4.588224e-03 0.0037000218 0.008281312 0.005759436 0.0053023528
#> A[T>A]T 4.534183e-03 0.0063134407 0.008168623 0.004858991 0.0082207520
#> C[T>A]A 1.845759e-03 0.0019581853 0.004404316 0.004088549 0.0018912552
#> C[T>A]C 5.806013e-03 0.0058919565 0.005116941 0.010057300 0.0009782504
#> C[T>A]G 3.947880e-03 0.0027266485 0.007617879 0.006518666 0.0043611914
#> C[T>A]T 4.697111e-03 0.0038426390 0.008445125 0.013041647 0.0037260357
#> G[T>A]A 1.446680e-03 0.0009450981 0.004410261 0.003061152 0.0026248922
#> G[T>A]C 2.986008e-03 0.0030922044 0.002966292 0.004141425 0.0015178545
#> G[T>A]G 2.934736e-03 0.0022516508 0.006013066 0.004922058 0.0027079864
#> G[T>A]T 2.129209e-03 0.0028520713 0.003729056 0.007605022 0.0007551720
#> T[T>A]A 4.648530e-03 0.0032625691 0.007419412 0.005524177 0.0065075505
#> T[T>A]C 1.090098e-02 0.0071819130 0.005562384 0.008627718 0.0015728777
#> T[T>A]G 3.647050e-03 0.0030843984 0.006244390 0.005778522 0.0017729856
#> T[T>A]T 7.062523e-03 0.0068424561 0.008551118 0.010322164 0.0025372244
#> A[T>C]A 4.077056e-02 0.0255635383 0.020695992 0.019644998 0.0184171121
#> A[T>C]C 2.387011e-02 0.0160341395 0.010453125 0.015635295 0.0048010126
#> A[T>C]G 4.022672e-02 0.0250080395 0.017320494 0.022140706 0.0120719020
#> A[T>C]T 2.456748e-02 0.0151623147 0.020012135 0.013704688 0.0108753289
#> C[T>C]A 2.519604e-02 0.0156092336 0.008568698 0.014062467 0.0050998648
#> C[T>C]C 2.793665e-02 0.0199829629 0.008792765 0.023098637 0.0027930397
#> C[T>C]G 3.480714e-02 0.0207201340 0.010599121 0.017721736 0.0080903070
#> C[T>C]T 2.613611e-02 0.0160280599 0.011228382 0.022852039 0.0072310190
#> G[T>C]A 4.509983e-02 0.0293680932 0.010316888 0.023151991 0.0062318446
#> G[T>C]C 2.052263e-02 0.0134891247 0.005331813 0.014128714 0.0035632117
#> G[T>C]G 3.199194e-02 0.0207411301 0.008485204 0.016728365 0.0037165519
#> G[T>C]T 3.237939e-02 0.0206522455 0.011616798 0.018375569 0.0069911470
#> T[T>C]A 4.024354e-02 0.0249039178 0.011677901 0.020122616 0.0059546613
#> T[T>C]C 2.744867e-02 0.0175283797 0.010181346 0.017269322 0.0046593827
#> T[T>C]G 2.499221e-02 0.0180551327 0.009008375 0.013324682 0.0066348593
#> T[T>C]T 3.101973e-02 0.0190799601 0.011272241 0.016995967 0.0049901055
#> A[T>G]A 1.738741e-03 0.0012891294 0.002447997 0.001647615 0.0019429212
#> A[T>G]C 5.603887e-04 0.0008272892 0.001636789 0.001698571 0.0018057899
#> A[T>G]G 1.540888e-03 0.0026056097 0.004063937 0.006023988 0.0042585765
#> A[T>G]T 2.340882e-03 0.0021415944 0.003399764 0.005635156 0.0032752150
#> C[T>G]A 1.299297e-03 0.0005943313 0.001753389 0.001478755 0.0006888606
#> C[T>G]C 3.882152e-03 0.0030504770 0.003076258 0.006460693 0.0026525892
#> C[T>G]G 4.278988e-03 0.0035171361 0.005262728 0.007513885 0.0044746952
#> C[T>G]T 2.768471e-03 0.0013521020 0.005078980 0.025166251 0.0036828507
#> G[T>G]A 3.134337e-04 0.0006051913 0.001501838 0.002323458 0.0008937109
#> G[T>G]C 1.581823e-05 0.0005654915 0.001052993 0.003423481 0.0003260169
#> G[T>G]G 1.283198e-03 0.0036426955 0.008106156 0.010937239 0.0030755801
#> G[T>G]T 1.938235e-03 0.0019914858 0.001949606 0.009740813 0.0012274704
#> T[T>G]A 2.239748e-03 0.0022685426 0.004545048 0.003739540 0.0027913751
#> T[T>G]C 2.753271e-03 0.0023656739 0.004788050 0.004909587 0.0030796233
#> T[T>G]G 1.702656e-03 0.0022183143 0.005969082 0.007596053 0.0034624082
#> T[T>G]T 2.839907e-03 0.0027864029 0.011268331 0.011579332 0.0103274820
#> Sig6 Sig7 Sig8 Sig9 Sig10
#> A[C>A]A 3.826092e-02 4.724913e-02 0.0178120539 0.0134224026 0.0120391543
#> A[C>A]C 3.836599e-02 8.241573e-03 0.0139272839 0.0033603340 0.0065572390
#> A[C>A]G 2.094793e-03 1.877803e-03 0.0012102579 0.0007943677 0.0015375507
#> A[C>A]T 3.641442e-02 1.588556e-02 0.0153677237 0.0065087852 0.0066336276
#> C[C>A]A 2.691809e-02 8.740133e-02 0.0131271236 0.0239850321 0.0155614286
#> C[C>A]C 2.733726e-02 1.811442e-02 0.0141389819 0.0052437058 0.0188099457
#> C[C>A]G 1.666300e-04 1.669183e-02 0.0024383188 0.0049664365 0.0068906550
#> C[C>A]T 2.765704e-02 3.358228e-02 0.0206655765 0.0087933355 0.0475854887
#> G[C>A]A 1.949879e-02 1.262603e-01 0.0070080108 0.0378816538 0.0127253347
#> G[C>A]C 1.538800e-02 1.438300e-02 0.0097578021 0.0053342788 0.0082831210
#> G[C>A]G 6.004324e-04 8.823974e-03 0.0017669051 0.0033750579 0.0028465644
#> G[C>A]T 1.999634e-02 6.981223e-02 0.0108119313 0.0216895840 0.0116465007
#> T[C>A]A 2.724107e-02 6.623871e-02 0.0064147522 0.0293409653 0.0129661113
#> T[C>A]C 1.582204e-02 4.164323e-02 0.0094472583 0.0153243751 0.0100660933
#> T[C>A]G 3.532825e-04 1.443802e-02 0.0021500240 0.0056357803 0.0014633689
#> T[C>A]T 3.517898e-02 1.157670e-01 0.0168354080 0.0376292317 0.0200493195
#> A[C>G]A 9.645533e-03 1.031817e-03 0.0146673491 0.0014754693 0.0080881830
#> A[C>G]C 5.135836e-03 2.209190e-03 0.0078484306 0.0022226966 0.0042084412
#> A[C>G]G 1.451011e-10 2.597836e-03 0.0027287740 0.0008169172 0.0019582682
#> A[C>G]T 7.897040e-03 2.668708e-03 0.0127185089 0.0032522025 0.0080256929
#> C[C>G]A 5.796642e-03 5.780703e-04 0.0086263852 0.0021906998 0.0033531303
#> C[C>G]C 5.310799e-03 2.752022e-03 0.0090260821 0.0017172840 0.0044565670
#> C[C>G]G 6.237546e-04 4.876815e-03 0.0018590308 0.0018933333 0.0012296529
#> C[C>G]T 6.274642e-03 5.001674e-03 0.0136617543 0.0040023620 0.0056625123
#> G[C>G]A 4.179727e-03 6.368407e-04 0.0059739128 0.0014569313 0.0031583787
#> G[C>G]C 3.233020e-03 1.317224e-04 0.0061827944 0.0008831625 0.0039056820
#> G[C>G]G 3.580418e-05 4.119368e-04 0.0008530248 0.0002924218 0.0003746795
#> G[C>G]T 6.478774e-03 1.438490e-03 0.0096749059 0.0017629033 0.0044325580
#> T[C>G]A 2.323334e-03 3.415215e-03 0.0060449161 0.0762079493 0.0022918252
#> T[C>G]C 1.698423e-03 7.502304e-03 0.0110975814 0.0194681430 0.0054747614
#> T[C>G]G 2.484306e-08 1.419331e-03 0.0012534141 0.0032769414 0.0008197758
#> T[C>G]T 1.846115e-03 9.024908e-03 0.0151944796 0.0888325266 0.0040744115
#> A[C>T]A 2.370786e-02 2.496054e-03 0.0176940316 0.0059672125 0.0258842538
#> A[C>T]C 6.582582e-03 1.919727e-03 0.0095584856 0.0033897338 0.0154495286
#> A[C>T]G 2.088552e-02 4.928558e-03 0.0222072201 0.0069633459 0.0321735112
#> A[C>T]T 2.253107e-02 3.322242e-03 0.0153794057 0.0042141857 0.0165521323
#> C[C>T]A 4.345598e-03 1.250457e-02 0.0168959988 0.0138731641 0.0129481390
#> C[C>T]C 3.165979e-03 5.384920e-03 0.0103419142 0.0033736149 0.0119390229
#> C[C>T]G 2.821050e-03 1.344941e-02 0.0227772235 0.0081081701 0.0440187948
#> C[C>T]T 2.145700e-02 6.152561e-03 0.0164242241 0.0070786229 0.0142299520
#> G[C>T]A 1.312104e-03 1.343438e-03 0.0220089101 0.0025389091 0.0464856158
#> G[C>T]C 2.315119e-05 1.748582e-03 0.0192937463 0.0004127408 0.0548388272
#> G[C>T]G 8.744178e-03 5.723040e-03 0.0300244012 0.0054582906 0.0714191409
#> G[C>T]T 6.928898e-03 1.529667e-03 0.0167362519 0.0005565121 0.0408701927
#> T[C>T]A 5.823772e-03 4.532300e-02 0.0442265088 0.0314452596 0.0070663450
#> T[C>T]C 2.253999e-03 1.611109e-02 0.0201997571 0.0120215042 0.0147789197
#> T[C>T]G 5.537855e-03 1.261871e-02 0.0148908088 0.0108637349 0.0157875262
#> T[C>T]T 9.974362e-03 2.076265e-02 0.0325808186 0.0110713497 0.0058688621
#> A[T>A]A 1.423292e-02 2.141479e-03 0.0074303693 0.0014631776 0.0066818314
#> A[T>A]C 1.272471e-02 6.333448e-04 0.0064094606 0.0012846173 0.0069751754
#> A[T>A]G 1.540660e-02 2.947117e-03 0.0077539962 0.0020676282 0.0058077176
#> A[T>A]T 3.217455e-02 1.523081e-04 0.0061796402 0.0026443216 0.0085698150
#> C[T>A]A 1.444728e-02 3.023481e-04 0.0062117635 0.0001486237 0.0026416320
#> C[T>A]C 2.100183e-02 2.090793e-03 0.0089687822 0.0053116300 0.0069589124
#> C[T>A]G 1.961117e-02 4.420913e-03 0.0083902941 0.0037940298 0.0075472520
#> C[T>A]T 2.846925e-02 1.744077e-03 0.0156355488 0.0162482758 0.0060410818
#> G[T>A]A 1.003547e-02 1.179375e-03 0.0045826999 0.0005598006 0.0051220725
#> G[T>A]C 8.060601e-03 1.189650e-03 0.0050306421 0.0014907148 0.0046399216
#> G[T>A]G 8.900403e-03 7.816584e-04 0.0071840138 0.0021044256 0.0040972843
#> G[T>A]T 2.037261e-02 3.045276e-03 0.0056529781 0.0034971232 0.0052962854
#> T[T>A]A 1.887312e-02 4.337576e-03 0.0061573573 0.0021505975 0.0038626313
#> T[T>A]C 1.175052e-02 1.169116e-03 0.0076919701 0.0027793934 0.0037798405
#> T[T>A]G 9.486536e-03 1.285751e-03 0.0045525439 0.0004788803 0.0042715468
#> T[T>A]T 2.992503e-02 2.695800e-03 0.0140448765 0.0022560741 0.0060524261
#> A[T>C]A 1.897613e-02 3.491014e-03 0.0116281383 0.0023708518 0.0201055777
#> A[T>C]C 9.644984e-03 1.144504e-03 0.0055507127 0.0061969947 0.0051800539
#> A[T>C]G 8.834625e-03 3.963776e-03 0.0108010017 0.0040994064 0.0232799206
#> A[T>C]T 2.660401e-02 3.479688e-03 0.0122898657 0.0036679653 0.0172913151
#> C[T>C]A 4.449973e-03 1.039728e-03 0.0061863797 0.0047714222 0.0065944432
#> C[T>C]C 7.738710e-03 3.462014e-03 0.0122715112 0.0204734083 0.0062898033
#> C[T>C]G 2.261935e-03 2.447112e-03 0.0121265701 0.0149242053 0.0228292536
#> C[T>C]T 1.011002e-02 2.729870e-03 0.0179000016 0.0509213996 0.0077815527
#> G[T>C]A 3.379184e-03 6.143812e-04 0.0045004640 0.0008852611 0.0089635145
#> G[T>C]C 6.094640e-03 2.476069e-03 0.0047404046 0.0056460031 0.0070417760
#> G[T>C]G 4.415338e-03 6.347855e-03 0.0038602710 0.0042234461 0.0099671842
#> G[T>C]T 4.797926e-03 5.018722e-03 0.0063903717 0.0055672133 0.0061755795
#> T[T>C]A 3.437703e-03 9.460714e-04 0.0049596680 0.0014353270 0.0071029725
#> T[T>C]C 5.192531e-03 6.050624e-03 0.0082631437 0.0074352610 0.0043747297
#> T[T>C]G 6.655287e-04 1.290089e-03 0.0033540732 0.0041272843 0.0073112724
#> T[T>C]T 9.884785e-03 1.394912e-03 0.0058716913 0.0048466513 0.0074672744
#> A[T>G]A 5.658475e-03 1.182289e-04 0.0014011203 0.0004073580 0.0025272667
#> A[T>G]C 8.513284e-04 1.784286e-03 0.0016842477 0.0038676952 0.0020974923
#> A[T>G]G 8.160365e-03 1.625460e-03 0.0055068951 0.0026325917 0.0032523391
#> A[T>G]T 2.151774e-03 2.087405e-03 0.0082282101 0.0158771327 0.0024145507
#> C[T>G]A 3.266457e-03 4.229457e-17 0.0015611360 0.0002631693 0.0006352990
#> C[T>G]C 1.932827e-03 1.637104e-03 0.0058766170 0.0099090014 0.0030969060
#> C[T>G]G 6.208937e-03 4.746647e-03 0.0077241759 0.0085753497 0.0068729791
#> C[T>G]T 1.801532e-03 5.301436e-03 0.0352045920 0.1309119350 0.0061794503
#> G[T>G]A 5.198928e-04 1.494047e-03 0.0023336045 0.0005736685 0.0010114515
#> G[T>G]C 1.696518e-04 3.541146e-05 0.0030584286 0.0047012463 0.0012350613
#> G[T>G]G 5.370159e-03 9.631609e-03 0.0083540934 0.0049040078 0.0041473985
#> G[T>G]T 4.921895e-03 3.192798e-03 0.0113362707 0.0304442125 0.0031258182
#> T[T>G]A 1.667187e-03 2.876156e-04 0.0033834647 0.0004168605 0.0031798650
#> T[T>G]C 2.507839e-03 2.692266e-03 0.0042889353 0.0055431476 0.0032710279
#> T[T>G]G 5.720718e-03 9.819975e-04 0.0078442218 0.0031388763 0.0033484695
#> T[T>G]T 9.261816e-03 9.452530e-04 0.0181123193 0.0275887115 0.0100161880
#> Sig11
#> A[C>A]A 0.0119643388
#> A[C>A]C 0.0113081129
#> A[C>A]G 0.0021554124
#> A[C>A]T 0.0095026398
#> C[C>A]A 0.0140018781
#> C[C>A]C 0.0112085274
#> C[C>A]G 0.0033745855
#> C[C>A]T 0.0216049082
#> G[C>A]A 0.0075259551
#> G[C>A]C 0.0086974062
#> G[C>A]G 0.0011124183
#> G[C>A]T 0.0088701419
#> T[C>A]A 0.0139543764
#> T[C>A]C 0.0131944655
#> T[C>A]G 0.0019420151
#> T[C>A]T 0.0177088881
#> A[C>G]A 0.0106766175
#> A[C>G]C 0.0056850823
#> A[C>G]G 0.0017416306
#> A[C>G]T 0.0107718592
#> C[C>G]A 0.0078516501
#> C[C>G]C 0.0056832804
#> C[C>G]G 0.0019173927
#> C[C>G]T 0.0100487174
#> G[C>G]A 0.0057315401
#> G[C>G]C 0.0031656594
#> G[C>G]G 0.0009532399
#> G[C>G]T 0.0079408169
#> T[C>G]A 0.0252838614
#> T[C>G]C 0.0100958922
#> T[C>G]G 0.0016943186
#> T[C>G]T 0.0427654345
#> A[C>T]A 0.0146486965
#> A[C>T]C 0.0077349315
#> A[C>T]G 0.0087236363
#> A[C>T]T 0.0140504467
#> C[C>T]A 0.0111932318
#> C[C>T]C 0.0059472256
#> C[C>T]G 0.0145907664
#> C[C>T]T 0.0085889933
#> G[C>T]A 0.0177498640
#> G[C>T]C 0.0163100076
#> G[C>T]G 0.0192857634
#> G[C>T]T 0.0151724714
#> T[C>T]A 0.1434999874
#> T[C>T]C 0.0329228076
#> T[C>T]G 0.0171585190
#> T[C>T]T 0.0815654393
#> A[T>A]A 0.0047901312
#> A[T>A]C 0.0065566317
#> A[T>A]G 0.0062986608
#> A[T>A]T 0.0078771656
#> C[T>A]A 0.0036529121
#> C[T>A]C 0.0074921966
#> C[T>A]G 0.0074136921
#> C[T>A]T 0.0079863388
#> G[T>A]A 0.0033487924
#> G[T>A]C 0.0029406654
#> G[T>A]G 0.0038110484
#> G[T>A]T 0.0043923215
#> T[T>A]A 0.0033251823
#> T[T>A]C 0.0062149076
#> T[T>A]G 0.0028027817
#> T[T>A]T 0.0097833133
#> A[T>C]A 0.0090339055
#> A[T>C]C 0.0079815971
#> A[T>C]G 0.0105328956
#> A[T>C]T 0.0102954236
#> C[T>C]A 0.0063542993
#> C[T>C]C 0.0079246661
#> C[T>C]G 0.0105837934
#> C[T>C]T 0.0055189942
#> G[T>C]A 0.0080367755
#> G[T>C]C 0.0046364642
#> G[T>C]G 0.0043811033
#> G[T>C]T 0.0057576598
#> T[T>C]A 0.0057357268
#> T[T>C]C 0.0081113805
#> T[T>C]G 0.0058331252
#> T[T>C]T 0.0073368347
#> A[T>G]A 0.0010862997
#> A[T>G]C 0.0010026306
#> A[T>G]G 0.0042593846
#> A[T>G]T 0.0031366896
#> C[T>G]A 0.0008762220
#> C[T>G]C 0.0029019742
#> C[T>G]G 0.0080639777
#> C[T>G]T 0.0004453111
#> G[T>G]A 0.0016244383
#> G[T>G]C 0.0022854326
#> G[T>G]G 0.0065617637
#> G[T>G]T 0.0032704409
#> T[T>G]A 0.0028479597
#> T[T>G]C 0.0042639236
#> T[T>G]G 0.0043744212
#> T[T>G]T 0.0069818933
#>
#> $Exposure
#> Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 Sample_7
#> Sig1 2892.6085 1438.31588 2144.9114 3.666354e+02 99.63291 137.893150
#> Sig2 3513.9127 1630.36643 2593.6703 5.099705e+02 35.87441 206.990635
#> Sig3 883.1682 999.82449 1620.5006 2.200093e+02 62.56996 136.248454
#> Sig4 613.7551 381.77322 1432.5917 3.561856e+01 61.76187 47.319553
#> Sig5 681.6584 996.24499 258.9395 5.827967e+00 84.75080 85.340279
#> Sig6 2736.6553 5.33738 173.6779 2.305192e-11 129.05447 8.009669
#> Sig7 772.0221 414.59774 518.4493 5.527642e+02 273.26348 326.195505
#> Sig8 1427.0286 961.67686 411.5051 1.636235e+02 212.96144 108.651649
#> Sig9 816.6163 563.91365 748.6491 2.449189e+02 44.50443 238.489037
#> Sig10 1867.2844 2427.52364 369.7361 2.104454e-11 227.89468 237.757576
#> Sig11 2619.2959 950.43175 313.4014 5.956370e+02 380.72101 64.108139
#> Sample_8 Sample_9 Sample_10 Sample_11 Sample_13 Sample_14 Sample_15
#> Sig1 89.93212 488.793291 905.4543 776.4751 348.97759 1016.18747 1460.2398
#> Sig2 31.35218 598.472660 472.9686 463.9699 479.85092 769.99088 1689.8349
#> Sig3 251.11465 861.183926 970.2283 720.3104 185.36715 355.95277 1666.7762
#> Sig4 108.58510 1235.038937 462.9401 119.2712 111.28595 196.68282 834.4780
#> Sig5 1009.90122 236.079620 436.4380 1091.2295 34.94721 145.36836 884.3407
#> Sig6 90.69113 60.886372 2410.8005 243.0323 273.73171 67.65131 310.1513
#> Sig7 440.48911 8.771324 1211.6532 130.4269 261.79152 225.22257 619.3689
#> Sig8 74.70018 1188.665460 938.4633 462.4500 75.00998 321.96583 766.1233
#> Sig9 79.56263 3007.538680 299.7510 167.8243 178.97157 160.12889 1782.2534
#> Sig10 57.18481 229.424132 464.3371 586.7409 50.61674 232.50772 572.2277
#> Sig11 158.47136 209.090714 1139.9931 494.2835 116.45055 719.36049 202.2660
#> Sample_16 Sample_18 Sample_20 Sample_21 Sample_23 Sample_25 Sample_26
#> Sig1 934.2772 160.1461 480.89236 686.2181 98.51086 209.63011 151.50822
#> Sig2 879.5054 340.8865 280.00483 1764.4183 26.83737 19.20814 334.19769
#> Sig3 755.1573 241.5888 93.86510 1550.0272 129.18451 265.97631 305.58491
#> Sig4 1081.1668 404.8437 187.53047 1562.8443 175.31600 442.56548 199.63635
#> Sig5 130.6072 284.1178 302.04813 385.5179 214.21455 1262.10468 1358.37761
#> Sig6 205.8966 1624.0754 71.94879 3375.6279 446.89891 27.27933 516.71080
#> Sig7 2249.0159 115.2477 703.74847 214.4457 1509.37124 3176.21408 18.33754
#> Sig8 230.3227 615.8694 109.10612 1382.4526 320.41983 929.48746 662.46444
#> Sig9 588.9454 1461.3680 264.78930 525.8701 764.31524 2347.32498 506.38697
#> Sig10 317.4797 178.5133 92.59464 690.8966 603.39723 714.40765 1536.45567
#> Sig11 353.5959 145.2997 10.46747 1193.6668 123.51717 767.71390 355.27864
#> Sample_27 Sample_28 Sample_29 Sample_30 Sample_1 Sample_12 Sample_17
#> Sig1 2128.2169 381.74682 441.29878 455.58574 6839.9396 7947.486 5275.766766
#> Sig2 2431.8323 147.91176 313.35221 316.96902 6459.2051 6858.115 3129.978687
#> Sig3 840.3103 286.50832 159.07111 21.13038 2722.7947 5464.118 1323.193417
#> Sig4 634.5028 206.81553 377.59857 245.44283 6487.6336 8892.051 2110.857319
#> Sig5 235.9003 186.55398 453.89135 150.34295 604.8369 1415.254 7304.314115
#> Sig6 1579.0797 194.60556 639.58753 560.70139 10107.1512 1325.049 4.146147
#> Sig7 667.4846 912.54872 66.25600 26.37378 952.4554 7312.888 559.804630
#> Sig8 408.5010 174.18293 384.03014 268.64216 3527.7995 2818.241 1671.982139
#> Sig9 604.7606 201.24096 709.88011 245.75308 1172.6965 2710.134 1397.714490
#> Sig10 257.2629 204.38831 54.05094 524.62055 1185.7381 1853.601 2977.616059
#> Sig11 302.2091 76.50792 48.95576 103.42140 2699.6698 3371.060 1916.421730
#> Sample_19 Sample_22 Sample_24
#> Sig1 5949.4895 4440.5748 5563.813
#> Sig2 3715.5190 1911.4240 7325.591
#> Sig3 3370.2751 6739.2437 7930.720
#> Sig4 2583.7245 3218.1267 4697.443
#> Sig5 4702.6021 7760.7238 2369.467
#> Sig6 1899.4088 2889.8528 2651.562
#> Sig7 5755.0451 419.3292 3705.740
#> Sig8 986.0466 4175.0721 3033.644
#> Sig9 1901.6070 727.1271 3134.564
#> Sig10 1259.3749 3983.8709 2613.616
#> Sig11 768.9564 2090.7489 2870.124
#>
#> $Exposure.norm
#> Sample_2 Sample_3 Sample_4 Sample_5 Sample_6 Sample_7
#> Sig1 0.15366594 0.1335482891 0.20261712 1.360426e-01 0.06176910 0.086344919
#> Sig2 0.18667189 0.1513802711 0.24500872 1.892280e-01 0.02224094 0.129611874
#> Sig3 0.04691712 0.0928341628 0.15307913 8.163593e-02 0.03879130 0.085315055
#> Sig4 0.03260491 0.0354478183 0.13532848 1.321651e-02 0.03829031 0.029630210
#> Sig5 0.03621218 0.0925018045 0.02446049 2.162507e-03 0.05254269 0.053437748
#> Sig6 0.14538114 0.0004955782 0.01640633 8.553571e-15 0.08000949 0.005015436
#> Sig7 0.04101264 0.0384955900 0.04897485 2.051069e-01 0.16941430 0.204254703
#> Sig8 0.07580898 0.0892921373 0.03887246 6.071361e-02 0.13202903 0.068034691
#> Sig9 0.04338164 0.0523596412 0.07072046 9.087881e-02 0.02759127 0.149335311
#> Sig10 0.09919697 0.2253966837 0.03492679 7.808719e-15 0.14128715 0.148877291
#> Sig11 0.13914658 0.0882480240 0.02960518 2.210151e-01 0.23603441 0.040142763
#> Sample_8 Sample_9 Sample_10 Sample_11 Sample_13 Sample_14
#> Sig1 0.03759728 0.060166986 0.09322060 0.14773080 0.16484527 0.24131628
#> Sig2 0.01310718 0.073667738 0.04869426 0.08827409 0.22666543 0.18285143
#> Sig3 0.10498172 0.106005631 0.09988938 0.13704499 0.08756121 0.08452889
#> Sig4 0.04539540 0.152024530 0.04766177 0.02269233 0.05256774 0.04670670
#> Sig5 0.42220225 0.029059726 0.04493326 0.20761541 0.01650789 0.03452094
#> Sig6 0.03791460 0.007494680 0.24820278 0.04623890 0.12930165 0.01606531
#> Sig7 0.18415216 0.001079688 0.12474517 0.02481480 0.12366151 0.05348410
#> Sig8 0.03122938 0.146316284 0.09661903 0.08798492 0.03543219 0.07645794
#> Sig9 0.03326219 0.370206671 0.03086071 0.03192996 0.08454015 0.03802616
#> Sig10 0.02390685 0.028240483 0.04780560 0.11163229 0.02390965 0.05521412
#> Sig11 0.06625100 0.025737583 0.11736744 0.09404151 0.05500732 0.17082812
#> Sample_15 Sample_16 Sample_18 Sample_20 Sample_21 Sample_23
#> Sig1 0.13535703 0.12092684 0.02874144 0.185172566 0.05147156 0.022328024
#> Sig2 0.15663936 0.11383754 0.06117896 0.107818750 0.13234475 0.006082837
#> Sig3 0.15450194 0.09774272 0.04335799 0.036143725 0.11626379 0.029280375
#> Sig4 0.07735200 0.13993929 0.07265737 0.072210544 0.11722517 0.039736328
#> Sig5 0.08197403 0.01690496 0.05099067 0.116306752 0.02891677 0.048552896
#> Sig6 0.02874950 0.02664993 0.29147310 0.027704626 0.25319769 0.101292076
#> Sig7 0.05741244 0.29109818 0.02068352 0.270985615 0.01608505 0.342107228
#> Sig8 0.07101585 0.02981150 0.11053019 0.042012437 0.10369443 0.072624903
#> Sig9 0.16520610 0.07622931 0.26227197 0.101959855 0.03944424 0.173236219
#> Sig10 0.05304269 0.04109254 0.03203781 0.035654522 0.05182248 0.136763274
#> Sig11 0.01874906 0.04576719 0.02607697 0.004030609 0.08953406 0.027995840
#> Sample_25 Sample_26 Sample_27 Sample_28 Sample_29 Sample_30
#> Sig1 0.020629002 0.025485244 0.21092211 0.12840412 0.12097097 0.156076860
#> Sig2 0.001890210 0.056215497 0.24101266 0.04975151 0.08589763 0.108588843
#> Sig3 0.026173844 0.051402532 0.08328100 0.09636975 0.04360535 0.007238951
#> Sig4 0.043551398 0.033580893 0.06288394 0.06956434 0.10350916 0.084085041
#> Sig5 0.124199527 0.228493118 0.02337948 0.06274918 0.12442291 0.051505245
#> Sig6 0.002684468 0.086916083 0.15649853 0.06545740 0.17532685 0.192087908
#> Sig7 0.312560672 0.003084564 0.06615268 0.30694430 0.01816242 0.009035262
#> Sig8 0.091467772 0.111433348 0.04048548 0.05858806 0.10527221 0.092032784
#> Sig9 0.230992450 0.085179509 0.05993627 0.06768928 0.19459580 0.084191330
#> Sig10 0.070302483 0.258447683 0.02549667 0.06874792 0.01481671 0.179727151
#> Sig11 0.075548174 0.059761530 0.02995117 0.02573415 0.01341999 0.035430625
#> Sample_1 Sample_12 Sample_17 Sample_19 Sample_22 Sample_24
#> Sig1 0.15996147 0.15905152 0.190655021 0.18087926 0.11577234 0.12122579
#> Sig2 0.15105746 0.13725016 0.113110791 0.11296101 0.04983365 0.15961185
#> Sig3 0.06367633 0.10935234 0.047817404 0.10246473 0.17570203 0.17279656
#> Sig4 0.15172230 0.17795493 0.076281907 0.07855164 0.08390131 0.10234909
#> Sig5 0.01414495 0.02832321 0.263962420 0.14297078 0.20233353 0.05162655
#> Sig6 0.23636974 0.02651795 0.000149833 0.05774675 0.07534273 0.05777290
#> Sig7 0.02227449 0.14635143 0.020230152 0.17496767 0.01093253 0.08074162
#> Sig8 0.08250248 0.05640091 0.060421888 0.02997827 0.10885029 0.06609781
#> Sig9 0.02742513 0.05423740 0.050510437 0.05781358 0.01895728 0.06829669
#> Sig10 0.02773013 0.03709576 0.107604729 0.03828813 0.10386540 0.05694613
#> Sig11 0.06313552 0.06746439 0.069255417 0.02337818 0.05450891 0.06253499
#>
#> $K
#> [1] 11
#>
#> attr(,"class")
#> [1] "Signature"
#> attr(,"used_runs")
#> [1] 6
#> attr(,"method")
#> [1] "brunet"
#> attr(,"call_method")
#> [1] "NMF with best practice"
#> attr(,"nrun")
#> [1] 50
#> attr(,"seed")
#> [1] 123456
# If you think the suggested signature number is not right
# Just pick up the solution you want
obj_s8 <- bp_get_sig_obj(e1, 8)
# Track the reconstructed profile similarity
rec_sim <- get_sig_rec_similarity(obj_s8, t(simulated_catalogs$set1))
rec_sim
#> sample similarity rss unexplained_variance
#> <char> <num> <num> <num>
#> 1: Sample_1 0.9997486 13230.30099 0.0005076483
#> 2: Sample_2 0.9992611 20030.77983 0.0014806239
#> 3: Sample_3 0.9903052 59263.95137 0.0196886410
#> 4: Sample_4 0.9997825 1856.76699 0.0004676679
#> 5: Sample_5 0.9992557 1019.54200 0.0015201050
#> 6: Sample_6 0.9958607 721.60675 0.0083609296
#> 7: Sample_7 0.9982163 261.63888 0.0036855218
#> 8: Sample_8 0.9801222 7122.56156 0.0410276351
#> 9: Sample_9 0.9992708 3331.53459 0.0014817779
#> 10: Sample_10 0.9989004 3735.70983 0.0022051099
#> 11: Sample_11 0.9993768 885.02187 0.0012459902
#> 12: Sample_12 0.9990641 77738.85426 0.0019042753
#> 13: Sample_13 0.9997332 87.78296 0.0005511132
#> 14: Sample_14 0.9995498 805.36015 0.0010261993
#> 15: Sample_15 0.9991823 5885.65491 0.0016497945
#> 16: Sample_16 0.9994722 1343.32088 0.0010669067
#> 17: Sample_17 0.9994158 24812.49173 0.0012580570
#> 18: Sample_18 0.9969664 4761.54340 0.0060580974
#> 19: Sample_19 0.9979704 77097.52442 0.0040672214
#> 20: Sample_20 0.9984978 397.72548 0.0030309129
#> 21: Sample_21 0.9995502 3540.17917 0.0009081402
#> 22: Sample_22 0.9996566 15566.63589 0.0006927896
#> 23: Sample_23 0.9973757 2453.12111 0.0052897034
#> 24: Sample_24 0.9999582 4645.77103 0.0000843955
#> 25: Sample_25 0.9996526 2058.53068 0.0006979333
#> 26: Sample_26 0.9950134 10587.56050 0.0100460865
#> 27: Sample_27 0.9992121 4574.39411 0.0015861921
#> 28: Sample_28 0.9983929 576.01938 0.0032494451
#> 29: Sample_29 0.9966799 1831.31566 0.0066974197
#> 30: Sample_30 0.9913698 2563.94388 0.0174160856
#> sample similarity rss unexplained_variance
#> <char> <num> <num> <num>
# After extraction, you can assign the signatures
# to reference COSMIC signatures
# More see ?get_sig_similarity
sim <- get_sig_similarity(obj_suggested)
#> -Comparing against COSMIC signatures
#> ------------------------------------
#> --Found Sig1 most similar to SBS12
#> Aetiology: Unknown [similarity: 0.827]
#> --Found Sig2 most similar to SBS13
#> Aetiology: Activity of APOBEC family of cytidine deaminases [similarity: 0.881]
#> --Found Sig3 most similar to SBS40
#> Aetiology: Unknown [similarity: 0.768]
#> --Found Sig4 most similar to SBS3
#> Aetiology: Defective homologous recombination DNA damage repair [similarity: 0.901]
#> --Found Sig5 most similar to SBS1
#> Aetiology: Spontaneous deamination of 5-methylcytosine (clock-like signature) [similarity: 0.917]
#> --Found Sig6 most similar to SBS8
#> Aetiology: Unknown [similarity: 0.939]
#> --Found Sig7 most similar to SBS18
#> Aetiology: Damage by reactive oxygen species [similarity: 0.975]
#> --Found Sig8 most similar to SBS40
#> Aetiology: Unknown [similarity: 0.849]
#> --Found Sig9 most similar to SBS17b
#> Aetiology: Unknown [similarity: 0.67]
#> --Found Sig10 most similar to SBS6
#> Aetiology: Defective DNA mismatch repair [similarity: 0.828]
#> --Found Sig11 most similar to SBS2
#> Aetiology: Activity of APOBEC family of cytidine deaminases [similarity: 0.872]
#> ------------------------------------
#> Return result invisiblely.
# Visualize the match result
if (require(pheatmap)) {
pheatmap::pheatmap(sim$similarity)
}
#> Loading required package: pheatmap
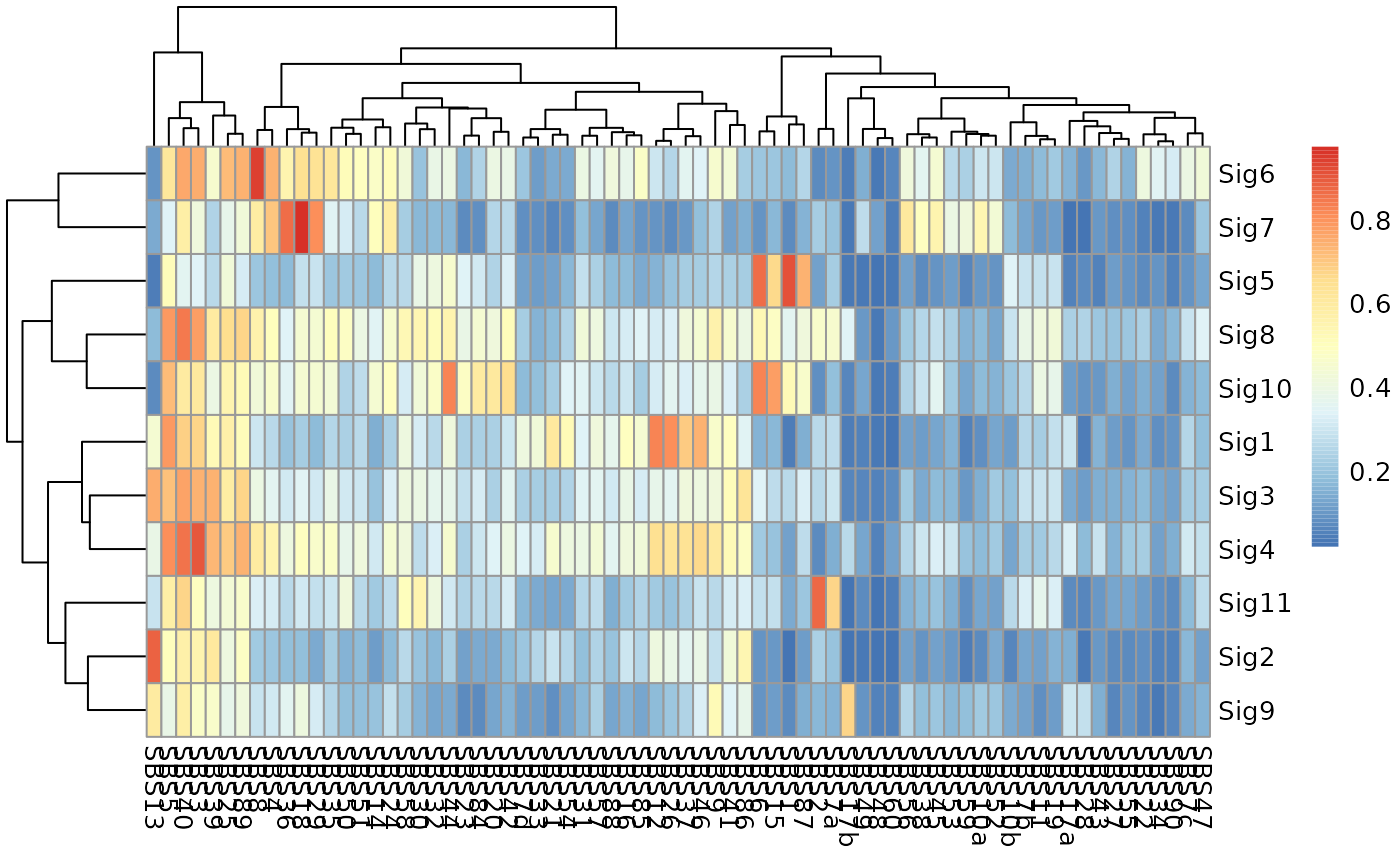 # You already got the activities of signatures
# in obj_suggested, however, you can still
# try to optimize the result.
# NOTE: the optimization step may not truly optimize the result!
expo <- bp_attribute_activity(e1, return_class = "data.table")
#> Set cache dir: /tmp/RtmpKIbCyr/sigminer_attribute_activity
#> Handling sample: Sample_2
#> ℹ [2025-12-25 15:35:42.955881]: Started.
#> ℹ [2025-12-25 15:35:42.958878]: Checking catalog.
#> ✔ [2025-12-25 15:35:42.961388]: Done.
#> ℹ [2025-12-25 15:35:42.963975]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:46.469824]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:46.472512]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:46.474877]: Done.
#> ℹ [2025-12-25 15:35:46.477314]: 3.521 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_2_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_2_sim.rds
#> Handling sample: Sample_3
#> ℹ [2025-12-25 15:35:46.706783]: Started.
#> ℹ [2025-12-25 15:35:46.709903]: Checking catalog.
#> ✔ [2025-12-25 15:35:46.712876]: Done.
#> ℹ [2025-12-25 15:35:46.715319]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:50.21962]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:50.222231]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:50.224647]: Done.
#> ℹ [2025-12-25 15:35:50.227078]: 3.52 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_3_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_3_sim.rds
#> Handling sample: Sample_4
#> ℹ [2025-12-25 15:35:50.43414]: Started.
#> ℹ [2025-12-25 15:35:50.437075]: Checking catalog.
#> ✔ [2025-12-25 15:35:50.439745]: Done.
#> ℹ [2025-12-25 15:35:50.4423]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:53.905439]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:53.908088]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:53.910525]: Done.
#> ℹ [2025-12-25 15:35:53.912963]: 3.479 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_4_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_4_sim.rds
#> Handling sample: Sample_5
#> ℹ [2025-12-25 15:35:54.129753]: Started.
#> ℹ [2025-12-25 15:35:54.132739]: Checking catalog.
#> ✔ [2025-12-25 15:35:54.13548]: Done.
#> ℹ [2025-12-25 15:35:54.138163]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:57.540606]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:57.543191]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:57.545621]: Done.
#> ℹ [2025-12-25 15:35:57.548034]: 3.418 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_5_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_5_sim.rds
#> Handling sample: Sample_6
#> ℹ [2025-12-25 15:35:57.753352]: Started.
#> ℹ [2025-12-25 15:35:57.756267]: Checking catalog.
#> ✔ [2025-12-25 15:35:57.758904]: Done.
#> ℹ [2025-12-25 15:35:57.761441]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:01.13361]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:01.136198]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:01.138632]: Done.
#> ℹ [2025-12-25 15:36:01.141048]: 3.388 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_6_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_6_sim.rds
#> Handling sample: Sample_7
#> ℹ [2025-12-25 15:36:01.346966]: Started.
#> ℹ [2025-12-25 15:36:01.349895]: Checking catalog.
#> ✔ [2025-12-25 15:36:01.352533]: Done.
#> ℹ [2025-12-25 15:36:01.355089]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:04.724263]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:04.726834]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:04.729485]: Done.
#> ℹ [2025-12-25 15:36:04.732039]: 3.385 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_7_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_7_sim.rds
#> Handling sample: Sample_8
#> ℹ [2025-12-25 15:36:04.938457]: Started.
#> ℹ [2025-12-25 15:36:04.941381]: Checking catalog.
#> ✔ [2025-12-25 15:36:04.944103]: Done.
#> ℹ [2025-12-25 15:36:04.946731]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:08.325742]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:08.32829]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:08.330669]: Done.
#> ℹ [2025-12-25 15:36:08.333063]: 3.395 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_8_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_8_sim.rds
#> Handling sample: Sample_9
#> ℹ [2025-12-25 15:36:08.539515]: Started.
#> ℹ [2025-12-25 15:36:08.542493]: Checking catalog.
#> ✔ [2025-12-25 15:36:08.545143]: Done.
#> ℹ [2025-12-25 15:36:08.547632]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:11.926575]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:11.92908]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:11.931393]: Done.
#> ℹ [2025-12-25 15:36:11.933745]: 3.394 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_9_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_9_sim.rds
#> Handling sample: Sample_10
#> ℹ [2025-12-25 15:36:12.144626]: Started.
#> ℹ [2025-12-25 15:36:12.147601]: Checking catalog.
#> ✔ [2025-12-25 15:36:12.150383]: Done.
#> ℹ [2025-12-25 15:36:12.152993]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:15.729772]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:15.732246]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:15.734512]: Done.
#> ℹ [2025-12-25 15:36:15.736836]: 3.592 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_10_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_10_sim.rds
#> Handling sample: Sample_11
#> ℹ [2025-12-25 15:36:15.945804]: Started.
#> ℹ [2025-12-25 15:36:15.948697]: Checking catalog.
#> ✔ [2025-12-25 15:36:15.951322]: Done.
#> ℹ [2025-12-25 15:36:15.953848]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:19.34714]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:19.349739]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:19.352102]: Done.
#> ℹ [2025-12-25 15:36:19.354566]: 3.409 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_11_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_11_sim.rds
#> Handling sample: Sample_13
#> ℹ [2025-12-25 15:36:19.561144]: Started.
#> ℹ [2025-12-25 15:36:19.563994]: Checking catalog.
#> ✔ [2025-12-25 15:36:19.566586]: Done.
#> ℹ [2025-12-25 15:36:19.569139]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:22.942522]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:22.94506]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:22.947376]: Done.
#> ℹ [2025-12-25 15:36:22.949811]: 3.389 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_13_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_13_sim.rds
#> Handling sample: Sample_14
#> ℹ [2025-12-25 15:36:23.155436]: Started.
#> ℹ [2025-12-25 15:36:23.158295]: Checking catalog.
#> ✔ [2025-12-25 15:36:23.160921]: Done.
#> ℹ [2025-12-25 15:36:23.163403]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:26.518328]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:26.52094]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:26.52335]: Done.
#> ℹ [2025-12-25 15:36:26.525917]: 3.37 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_14_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_14_sim.rds
#> Handling sample: Sample_15
#> ℹ [2025-12-25 15:36:26.731966]: Started.
#> ℹ [2025-12-25 15:36:26.734815]: Checking catalog.
#> ✔ [2025-12-25 15:36:26.737427]: Done.
#> ℹ [2025-12-25 15:36:26.740029]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:30.126943]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:30.129418]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:30.131733]: Done.
#> ℹ [2025-12-25 15:36:30.134082]: 3.402 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_15_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_15_sim.rds
#> Handling sample: Sample_16
#> ℹ [2025-12-25 15:36:30.336756]: Started.
#> ℹ [2025-12-25 15:36:30.339568]: Checking catalog.
#> ✔ [2025-12-25 15:36:30.342208]: Done.
#> ℹ [2025-12-25 15:36:30.344699]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:33.690935]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:33.693389]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:33.695662]: Done.
#> ℹ [2025-12-25 15:36:33.698039]: 3.361 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_16_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_16_sim.rds
#> Handling sample: Sample_18
#> ℹ [2025-12-25 15:36:33.897796]: Started.
#> ℹ [2025-12-25 15:36:33.900597]: Checking catalog.
#> ✔ [2025-12-25 15:36:33.903209]: Done.
#> ℹ [2025-12-25 15:36:33.905716]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:37.216583]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:37.219131]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:37.221428]: Done.
#> ℹ [2025-12-25 15:36:37.223754]: 3.326 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_18_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_18_sim.rds
#> Handling sample: Sample_20
#> ℹ [2025-12-25 15:36:37.436796]: Started.
#> ℹ [2025-12-25 15:36:37.4394]: Checking catalog.
#> ✔ [2025-12-25 15:36:37.441725]: Done.
#> ℹ [2025-12-25 15:36:37.443986]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:40.782493]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:40.785017]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:40.787312]: Done.
#> ℹ [2025-12-25 15:36:40.789661]: 3.353 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_20_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_20_sim.rds
#> Handling sample: Sample_21
#> ℹ [2025-12-25 15:36:41.000819]: Started.
#> ℹ [2025-12-25 15:36:41.0037]: Checking catalog.
#> ✔ [2025-12-25 15:36:41.006346]: Done.
#> ℹ [2025-12-25 15:36:41.008875]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:44.555591]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:44.558049]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:44.560288]: Done.
#> ℹ [2025-12-25 15:36:44.562596]: 3.562 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_21_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_21_sim.rds
#> Handling sample: Sample_23
#> ℹ [2025-12-25 15:36:44.766339]: Started.
#> ℹ [2025-12-25 15:36:44.769553]: Checking catalog.
#> ✔ [2025-12-25 15:36:44.772241]: Done.
#> ℹ [2025-12-25 15:36:44.774771]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:48.098048]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:48.100516]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:48.10282]: Done.
#> ℹ [2025-12-25 15:36:48.105154]: 3.339 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_23_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_23_sim.rds
#> Handling sample: Sample_25
#> ℹ [2025-12-25 15:36:48.308407]: Started.
#> ℹ [2025-12-25 15:36:48.311222]: Checking catalog.
#> ✔ [2025-12-25 15:36:48.31379]: Done.
#> ℹ [2025-12-25 15:36:48.316283]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:51.673909]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:51.676357]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:51.678725]: Done.
#> ℹ [2025-12-25 15:36:51.681066]: 3.373 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_25_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_25_sim.rds
#> Handling sample: Sample_26
#> ℹ [2025-12-25 15:36:51.880134]: Started.
#> ℹ [2025-12-25 15:36:51.882952]: Checking catalog.
#> ✔ [2025-12-25 15:36:51.885531]: Done.
#> ℹ [2025-12-25 15:36:51.88801]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:55.216022]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:55.218521]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:55.220863]: Done.
#> ℹ [2025-12-25 15:36:55.223279]: 3.343 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_26_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_26_sim.rds
#> Handling sample: Sample_27
#> ℹ [2025-12-25 15:36:55.427512]: Started.
#> ℹ [2025-12-25 15:36:55.430399]: Checking catalog.
#> ✔ [2025-12-25 15:36:55.433026]: Done.
#> ℹ [2025-12-25 15:36:55.435473]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:58.77934]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:58.781945]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:58.784264]: Done.
#> ℹ [2025-12-25 15:36:58.786629]: 3.359 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_27_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_27_sim.rds
#> Handling sample: Sample_28
#> ℹ [2025-12-25 15:36:58.98665]: Started.
#> ℹ [2025-12-25 15:36:58.989491]: Checking catalog.
#> ✔ [2025-12-25 15:36:58.992096]: Done.
#> ℹ [2025-12-25 15:36:58.994581]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:02.279981]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:02.282409]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:02.284716]: Done.
#> ℹ [2025-12-25 15:37:02.287018]: 3.3 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_28_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_28_sim.rds
#> Handling sample: Sample_29
#> ℹ [2025-12-25 15:37:02.493197]: Started.
#> ℹ [2025-12-25 15:37:02.496031]: Checking catalog.
#> ✔ [2025-12-25 15:37:02.498611]: Done.
#> ℹ [2025-12-25 15:37:02.501108]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:05.801925]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:05.804452]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:05.80687]: Done.
#> ℹ [2025-12-25 15:37:05.80936]: 3.316 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_29_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_29_sim.rds
#> Handling sample: Sample_30
#> ℹ [2025-12-25 15:37:06.019817]: Started.
#> ℹ [2025-12-25 15:37:06.022622]: Checking catalog.
#> ✔ [2025-12-25 15:37:06.02522]: Done.
#> ℹ [2025-12-25 15:37:06.027735]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:09.333281]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:09.335829]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:09.338178]: Done.
#> ℹ [2025-12-25 15:37:09.340587]: 3.321 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_30_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_30_sim.rds
#> Handling sample: Sample_1
#> ℹ [2025-12-25 15:37:09.545791]: Started.
#> ℹ [2025-12-25 15:37:09.5486]: Checking catalog.
#> ✔ [2025-12-25 15:37:09.55125]: Done.
#> ℹ [2025-12-25 15:37:09.553802]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:13.315743]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:13.318196]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:13.320461]: Done.
#> ℹ [2025-12-25 15:37:13.322822]: 3.777 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_1_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_1_sim.rds
#> Handling sample: Sample_12
#> ℹ [2025-12-25 15:37:13.526856]: Started.
#> ℹ [2025-12-25 15:37:13.529734]: Checking catalog.
#> ✔ [2025-12-25 15:37:13.532355]: Done.
#> ℹ [2025-12-25 15:37:13.534907]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:17.168985]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:17.171494]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:17.173981]: Done.
#> ℹ [2025-12-25 15:37:17.176415]: 3.65 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_12_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_12_sim.rds
#> Handling sample: Sample_17
#> ℹ [2025-12-25 15:37:17.38101]: Started.
#> ℹ [2025-12-25 15:37:17.38393]: Checking catalog.
#> ✔ [2025-12-25 15:37:17.386547]: Done.
#> ℹ [2025-12-25 15:37:17.388844]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:20.87149]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:20.874344]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:20.87697]: Done.
#> ℹ [2025-12-25 15:37:20.879473]: 3.498 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_17_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_17_sim.rds
#> Handling sample: Sample_19
#> ℹ [2025-12-25 15:37:21.087471]: Started.
#> ℹ [2025-12-25 15:37:21.090353]: Checking catalog.
#> ✔ [2025-12-25 15:37:21.093003]: Done.
#> ℹ [2025-12-25 15:37:21.095426]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:24.596549]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:24.599072]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:24.601451]: Done.
#> ℹ [2025-12-25 15:37:24.603854]: 3.516 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_19_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_19_sim.rds
#> Handling sample: Sample_22
#> ℹ [2025-12-25 15:37:24.804303]: Started.
#> ℹ [2025-12-25 15:37:24.807194]: Checking catalog.
#> ✔ [2025-12-25 15:37:24.809925]: Done.
#> ℹ [2025-12-25 15:37:24.812411]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:28.334829]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:28.337293]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:28.339576]: Done.
#> ℹ [2025-12-25 15:37:28.341899]: 3.538 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_22_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_22_sim.rds
#> Handling sample: Sample_24
#> ℹ [2025-12-25 15:37:28.547782]: Started.
#> ℹ [2025-12-25 15:37:28.550613]: Checking catalog.
#> ✔ [2025-12-25 15:37:28.55327]: Done.
#> ℹ [2025-12-25 15:37:28.555826]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:32.150843]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:32.153289]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:32.155722]: Done.
#> ℹ [2025-12-25 15:37:32.158028]: 3.61 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_24_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_24_sim.rds
#>
#> Reading result files...
#> Clean cache files when exits.
#> Done.
expo$abs_activity
#> sample Sig1 Sig2 Sig3 Sig4 Sig5
#> <char> <num> <num> <num> <num> <num>
#> 1: Sample_2 0.0000 8324.3129 0.00000 0.00000 836.23229
#> 2: Sample_3 0.0000 5134.2448 0.00000 0.00000 1711.02871
#> 3: Sample_4 0.0000 7628.7041 1523.89284 0.00000 0.00000
#> 4: Sample_5 0.0000 1033.6448 0.00000 0.00000 0.00000
#> 5: Sample_6 0.0000 0.0000 0.00000 0.00000 181.69335
#> 6: Sample_7 0.0000 582.8461 0.00000 0.00000 172.01602
#> 7: Sample_8 0.0000 0.0000 0.00000 0.00000 1369.45498
#> 8: Sample_9 0.0000 0.0000 0.00000 0.00000 0.00000
#> 9: Sample_10 1143.1114 0.0000 0.00000 0.00000 0.00000
#> 10: Sample_11 0.0000 1432.9232 0.00000 0.00000 1600.13559
#> 11: Sample_13 0.0000 1570.0646 0.00000 0.00000 56.91541
#> 12: Sample_14 0.0000 1331.7105 0.00000 0.00000 0.00000
#> 13: Sample_15 0.0000 3713.2011 2380.78415 0.00000 551.04363
#> 14: Sample_16 673.5523 1095.9283 0.00000 2397.98433 0.00000
#> 15: Sample_18 0.0000 0.0000 0.00000 0.00000 178.07434
#> 16: Sample_20 440.5282 0.0000 0.00000 822.08119 416.87212
#> 17: Sample_21 0.0000 3630.3894 4917.63789 0.00000 0.00000
#> 18: Sample_23 0.0000 0.0000 0.00000 33.20335 243.22641
#> 19: Sample_25 0.0000 0.0000 0.00000 0.00000 2141.32871
#> 20: Sample_26 0.0000 429.4069 0.00000 0.00000 2491.68582
#> 21: Sample_27 0.0000 6699.1632 76.18949 0.00000 194.17338
#> 22: Sample_28 302.2635 0.0000 0.00000 818.94743 0.00000
#> 23: Sample_29 0.0000 0.0000 0.00000 1097.81229 578.42602
#> 24: Sample_30 400.9180 0.0000 0.00000 826.83399 238.78021
#> 25: Sample_1 9138.4848 0.0000 0.00000 18252.04649 0.00000
#> 26: Sample_12 8847.7116 2860.9798 5597.61520 18490.61967 0.00000
#> 27: Sample_17 8282.3148 0.0000 0.00000 0.00000 12385.06731
#> 28: Sample_19 10367.8524 1558.8368 0.00000 4600.13526 5734.80507
#> 29: Sample_22 3572.9161 0.0000 0.00000 11095.51018 8944.23850
#> 30: Sample_24 0.0000 15548.7912 22224.83696 0.00000 0.00000
#> sample Sig1 Sig2 Sig3 Sig4 Sig5
#> <char> <num> <num> <num> <num> <num>
#> Sig6 Sig7 Sig8 Sig9 Sig10 Sig11
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.00000 0.0000 0.0000 0.00000 0.0000 9633.1075
#> 2: 0.00000 199.5056 0.0000 247.33530 3412.1065 0.0000
#> 3: 0.00000 1005.7703 0.0000 330.72220 0.0000 135.6430
#> 4: 0.00000 0.0000 0.0000 0.00000 0.0000 1661.3552
#> 5: 0.00000 226.9018 0.0000 0.00000 0.0000 1197.6510
#> 6: 0.00000 307.6999 0.0000 367.55856 149.6321 0.0000
#> 7: 0.00000 482.2529 0.0000 0.00000 0.0000 537.6053
#> 8: 0.00000 0.0000 2783.9792 5340.02083 0.0000 0.0000
#> 9: 2541.17025 1129.0811 1701.7448 0.00000 0.0000 3136.6450
#> 10: 0.00000 0.0000 0.0000 81.98904 192.4109 1940.1209
#> 11: 13.19321 401.8427 0.0000 46.59928 0.0000 0.0000
#> 12: 0.00000 0.0000 0.0000 0.00000 0.0000 2879.2895
#> 13: 0.00000 0.0000 0.0000 3589.13531 0.0000 524.3026
#> 14: 430.87300 2892.9436 0.0000 0.00000 238.9572 0.0000
#> 15: 405.92264 0.0000 2134.3934 2835.14250 0.0000 0.0000
#> 16: 40.38733 728.0084 0.0000 0.00000 148.8330 0.0000
#> 17: 3540.94597 502.9650 0.0000 561.13782 0.0000 0.0000
#> 18: 532.02063 1582.7356 0.0000 847.56904 1161.9916 0.0000
#> 19: 0.00000 2796.0676 1904.0547 2645.43897 0.0000 698.4211
#> 20: 0.00000 0.0000 0.0000 926.44717 2092.5266 0.0000
#> 21: 1587.79622 906.3852 0.0000 212.54317 0.0000 0.0000
#> 22: 362.25152 1080.0412 0.0000 0.00000 402.3160 0.0000
#> 23: 176.34483 0.0000 965.4548 830.89664 0.0000 0.0000
#> 24: 253.43957 0.0000 223.5043 100.89195 882.4947 0.0000
#> 25: 13782.50908 0.0000 0.0000 0.00000 1493.6083 0.0000
#> 26: 3816.65478 9141.6032 0.0000 0.00000 1195.2922 0.0000
#> 27: 0.00000 0.0000 1629.9891 0.00000 2218.2940 3163.1918
#> 28: 1724.13326 7400.3912 0.0000 0.00000 1469.2849 0.0000
#> 29: 5109.09700 0.0000 3655.6147 0.00000 6016.8222 0.0000
#> 30: 679.65994 5732.9665 0.0000 1757.38556 0.0000 0.0000
#> Sig6 Sig7 Sig8 Sig9 Sig10 Sig11
#> <num> <num> <num> <num> <num> <num>
# }
if (FALSE) { # \dontrun{
# Iterative extraction:
# This procedure will rerun extraction step
# for those samples with reconstructed catalog similarity
# lower than a threshold (default is 0.95)
e2 <- bp_extract_signatures_iter(
t(simulated_catalogs$set1),
range = 9:11,
n_bootstrap = 5,
n_nmf_run = 5,
sim_threshold = 0.99
)
e2
# When the procedure run multiple rounds
# you can cluster the signatures from different rounds by
# the following command
# bp_cluster_iter_list(e2)
## Extra utilities
rank_score <- bp_get_rank_score(e1)
rank_score
stats <- bp_get_stats(e2$iter1)
# Get the mean reconstructed similarity
1 - stats$stats_sample$cosine_distance_mean
} # }
# You already got the activities of signatures
# in obj_suggested, however, you can still
# try to optimize the result.
# NOTE: the optimization step may not truly optimize the result!
expo <- bp_attribute_activity(e1, return_class = "data.table")
#> Set cache dir: /tmp/RtmpKIbCyr/sigminer_attribute_activity
#> Handling sample: Sample_2
#> ℹ [2025-12-25 15:35:42.955881]: Started.
#> ℹ [2025-12-25 15:35:42.958878]: Checking catalog.
#> ✔ [2025-12-25 15:35:42.961388]: Done.
#> ℹ [2025-12-25 15:35:42.963975]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:46.469824]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:46.472512]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:46.474877]: Done.
#> ℹ [2025-12-25 15:35:46.477314]: 3.521 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_2_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_2_sim.rds
#> Handling sample: Sample_3
#> ℹ [2025-12-25 15:35:46.706783]: Started.
#> ℹ [2025-12-25 15:35:46.709903]: Checking catalog.
#> ✔ [2025-12-25 15:35:46.712876]: Done.
#> ℹ [2025-12-25 15:35:46.715319]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:50.21962]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:50.222231]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:50.224647]: Done.
#> ℹ [2025-12-25 15:35:50.227078]: 3.52 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_3_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_3_sim.rds
#> Handling sample: Sample_4
#> ℹ [2025-12-25 15:35:50.43414]: Started.
#> ℹ [2025-12-25 15:35:50.437075]: Checking catalog.
#> ✔ [2025-12-25 15:35:50.439745]: Done.
#> ℹ [2025-12-25 15:35:50.4423]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:53.905439]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:53.908088]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:53.910525]: Done.
#> ℹ [2025-12-25 15:35:53.912963]: 3.479 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_4_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_4_sim.rds
#> Handling sample: Sample_5
#> ℹ [2025-12-25 15:35:54.129753]: Started.
#> ℹ [2025-12-25 15:35:54.132739]: Checking catalog.
#> ✔ [2025-12-25 15:35:54.13548]: Done.
#> ℹ [2025-12-25 15:35:54.138163]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:35:57.540606]: Signature exposures collected.
#> ✔ [2025-12-25 15:35:57.543191]: Errors and similarity collected.
#> ✔ [2025-12-25 15:35:57.545621]: Done.
#> ℹ [2025-12-25 15:35:57.548034]: 3.418 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_5_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_5_sim.rds
#> Handling sample: Sample_6
#> ℹ [2025-12-25 15:35:57.753352]: Started.
#> ℹ [2025-12-25 15:35:57.756267]: Checking catalog.
#> ✔ [2025-12-25 15:35:57.758904]: Done.
#> ℹ [2025-12-25 15:35:57.761441]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:01.13361]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:01.136198]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:01.138632]: Done.
#> ℹ [2025-12-25 15:36:01.141048]: 3.388 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_6_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_6_sim.rds
#> Handling sample: Sample_7
#> ℹ [2025-12-25 15:36:01.346966]: Started.
#> ℹ [2025-12-25 15:36:01.349895]: Checking catalog.
#> ✔ [2025-12-25 15:36:01.352533]: Done.
#> ℹ [2025-12-25 15:36:01.355089]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:04.724263]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:04.726834]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:04.729485]: Done.
#> ℹ [2025-12-25 15:36:04.732039]: 3.385 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_7_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_7_sim.rds
#> Handling sample: Sample_8
#> ℹ [2025-12-25 15:36:04.938457]: Started.
#> ℹ [2025-12-25 15:36:04.941381]: Checking catalog.
#> ✔ [2025-12-25 15:36:04.944103]: Done.
#> ℹ [2025-12-25 15:36:04.946731]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:08.325742]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:08.32829]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:08.330669]: Done.
#> ℹ [2025-12-25 15:36:08.333063]: 3.395 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_8_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_8_sim.rds
#> Handling sample: Sample_9
#> ℹ [2025-12-25 15:36:08.539515]: Started.
#> ℹ [2025-12-25 15:36:08.542493]: Checking catalog.
#> ✔ [2025-12-25 15:36:08.545143]: Done.
#> ℹ [2025-12-25 15:36:08.547632]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:11.926575]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:11.92908]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:11.931393]: Done.
#> ℹ [2025-12-25 15:36:11.933745]: 3.394 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_9_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_9_sim.rds
#> Handling sample: Sample_10
#> ℹ [2025-12-25 15:36:12.144626]: Started.
#> ℹ [2025-12-25 15:36:12.147601]: Checking catalog.
#> ✔ [2025-12-25 15:36:12.150383]: Done.
#> ℹ [2025-12-25 15:36:12.152993]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:15.729772]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:15.732246]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:15.734512]: Done.
#> ℹ [2025-12-25 15:36:15.736836]: 3.592 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_10_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_10_sim.rds
#> Handling sample: Sample_11
#> ℹ [2025-12-25 15:36:15.945804]: Started.
#> ℹ [2025-12-25 15:36:15.948697]: Checking catalog.
#> ✔ [2025-12-25 15:36:15.951322]: Done.
#> ℹ [2025-12-25 15:36:15.953848]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:19.34714]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:19.349739]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:19.352102]: Done.
#> ℹ [2025-12-25 15:36:19.354566]: 3.409 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_11_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_11_sim.rds
#> Handling sample: Sample_13
#> ℹ [2025-12-25 15:36:19.561144]: Started.
#> ℹ [2025-12-25 15:36:19.563994]: Checking catalog.
#> ✔ [2025-12-25 15:36:19.566586]: Done.
#> ℹ [2025-12-25 15:36:19.569139]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:22.942522]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:22.94506]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:22.947376]: Done.
#> ℹ [2025-12-25 15:36:22.949811]: 3.389 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_13_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_13_sim.rds
#> Handling sample: Sample_14
#> ℹ [2025-12-25 15:36:23.155436]: Started.
#> ℹ [2025-12-25 15:36:23.158295]: Checking catalog.
#> ✔ [2025-12-25 15:36:23.160921]: Done.
#> ℹ [2025-12-25 15:36:23.163403]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:26.518328]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:26.52094]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:26.52335]: Done.
#> ℹ [2025-12-25 15:36:26.525917]: 3.37 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_14_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_14_sim.rds
#> Handling sample: Sample_15
#> ℹ [2025-12-25 15:36:26.731966]: Started.
#> ℹ [2025-12-25 15:36:26.734815]: Checking catalog.
#> ✔ [2025-12-25 15:36:26.737427]: Done.
#> ℹ [2025-12-25 15:36:26.740029]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:30.126943]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:30.129418]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:30.131733]: Done.
#> ℹ [2025-12-25 15:36:30.134082]: 3.402 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_15_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_15_sim.rds
#> Handling sample: Sample_16
#> ℹ [2025-12-25 15:36:30.336756]: Started.
#> ℹ [2025-12-25 15:36:30.339568]: Checking catalog.
#> ✔ [2025-12-25 15:36:30.342208]: Done.
#> ℹ [2025-12-25 15:36:30.344699]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:33.690935]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:33.693389]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:33.695662]: Done.
#> ℹ [2025-12-25 15:36:33.698039]: 3.361 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_16_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_16_sim.rds
#> Handling sample: Sample_18
#> ℹ [2025-12-25 15:36:33.897796]: Started.
#> ℹ [2025-12-25 15:36:33.900597]: Checking catalog.
#> ✔ [2025-12-25 15:36:33.903209]: Done.
#> ℹ [2025-12-25 15:36:33.905716]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:37.216583]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:37.219131]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:37.221428]: Done.
#> ℹ [2025-12-25 15:36:37.223754]: 3.326 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_18_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_18_sim.rds
#> Handling sample: Sample_20
#> ℹ [2025-12-25 15:36:37.436796]: Started.
#> ℹ [2025-12-25 15:36:37.4394]: Checking catalog.
#> ✔ [2025-12-25 15:36:37.441725]: Done.
#> ℹ [2025-12-25 15:36:37.443986]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:40.782493]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:40.785017]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:40.787312]: Done.
#> ℹ [2025-12-25 15:36:40.789661]: 3.353 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_20_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_20_sim.rds
#> Handling sample: Sample_21
#> ℹ [2025-12-25 15:36:41.000819]: Started.
#> ℹ [2025-12-25 15:36:41.0037]: Checking catalog.
#> ✔ [2025-12-25 15:36:41.006346]: Done.
#> ℹ [2025-12-25 15:36:41.008875]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:44.555591]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:44.558049]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:44.560288]: Done.
#> ℹ [2025-12-25 15:36:44.562596]: 3.562 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_21_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_21_sim.rds
#> Handling sample: Sample_23
#> ℹ [2025-12-25 15:36:44.766339]: Started.
#> ℹ [2025-12-25 15:36:44.769553]: Checking catalog.
#> ✔ [2025-12-25 15:36:44.772241]: Done.
#> ℹ [2025-12-25 15:36:44.774771]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:48.098048]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:48.100516]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:48.10282]: Done.
#> ℹ [2025-12-25 15:36:48.105154]: 3.339 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_23_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_23_sim.rds
#> Handling sample: Sample_25
#> ℹ [2025-12-25 15:36:48.308407]: Started.
#> ℹ [2025-12-25 15:36:48.311222]: Checking catalog.
#> ✔ [2025-12-25 15:36:48.31379]: Done.
#> ℹ [2025-12-25 15:36:48.316283]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:51.673909]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:51.676357]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:51.678725]: Done.
#> ℹ [2025-12-25 15:36:51.681066]: 3.373 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_25_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_25_sim.rds
#> Handling sample: Sample_26
#> ℹ [2025-12-25 15:36:51.880134]: Started.
#> ℹ [2025-12-25 15:36:51.882952]: Checking catalog.
#> ✔ [2025-12-25 15:36:51.885531]: Done.
#> ℹ [2025-12-25 15:36:51.88801]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:55.216022]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:55.218521]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:55.220863]: Done.
#> ℹ [2025-12-25 15:36:55.223279]: 3.343 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_26_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_26_sim.rds
#> Handling sample: Sample_27
#> ℹ [2025-12-25 15:36:55.427512]: Started.
#> ℹ [2025-12-25 15:36:55.430399]: Checking catalog.
#> ✔ [2025-12-25 15:36:55.433026]: Done.
#> ℹ [2025-12-25 15:36:55.435473]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:36:58.77934]: Signature exposures collected.
#> ✔ [2025-12-25 15:36:58.781945]: Errors and similarity collected.
#> ✔ [2025-12-25 15:36:58.784264]: Done.
#> ℹ [2025-12-25 15:36:58.786629]: 3.359 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_27_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_27_sim.rds
#> Handling sample: Sample_28
#> ℹ [2025-12-25 15:36:58.98665]: Started.
#> ℹ [2025-12-25 15:36:58.989491]: Checking catalog.
#> ✔ [2025-12-25 15:36:58.992096]: Done.
#> ℹ [2025-12-25 15:36:58.994581]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:02.279981]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:02.282409]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:02.284716]: Done.
#> ℹ [2025-12-25 15:37:02.287018]: 3.3 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_28_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_28_sim.rds
#> Handling sample: Sample_29
#> ℹ [2025-12-25 15:37:02.493197]: Started.
#> ℹ [2025-12-25 15:37:02.496031]: Checking catalog.
#> ✔ [2025-12-25 15:37:02.498611]: Done.
#> ℹ [2025-12-25 15:37:02.501108]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:05.801925]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:05.804452]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:05.80687]: Done.
#> ℹ [2025-12-25 15:37:05.80936]: 3.316 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_29_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_29_sim.rds
#> Handling sample: Sample_30
#> ℹ [2025-12-25 15:37:06.019817]: Started.
#> ℹ [2025-12-25 15:37:06.022622]: Checking catalog.
#> ✔ [2025-12-25 15:37:06.02522]: Done.
#> ℹ [2025-12-25 15:37:06.027735]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:09.333281]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:09.335829]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:09.338178]: Done.
#> ℹ [2025-12-25 15:37:09.340587]: 3.321 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_30_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_30_sim.rds
#> Handling sample: Sample_1
#> ℹ [2025-12-25 15:37:09.545791]: Started.
#> ℹ [2025-12-25 15:37:09.5486]: Checking catalog.
#> ✔ [2025-12-25 15:37:09.55125]: Done.
#> ℹ [2025-12-25 15:37:09.553802]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:13.315743]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:13.318196]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:13.320461]: Done.
#> ℹ [2025-12-25 15:37:13.322822]: 3.777 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_1_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_1_sim.rds
#> Handling sample: Sample_12
#> ℹ [2025-12-25 15:37:13.526856]: Started.
#> ℹ [2025-12-25 15:37:13.529734]: Checking catalog.
#> ✔ [2025-12-25 15:37:13.532355]: Done.
#> ℹ [2025-12-25 15:37:13.534907]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:17.168985]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:17.171494]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:17.173981]: Done.
#> ℹ [2025-12-25 15:37:17.176415]: 3.65 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_12_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_12_sim.rds
#> Handling sample: Sample_17
#> ℹ [2025-12-25 15:37:17.38101]: Started.
#> ℹ [2025-12-25 15:37:17.38393]: Checking catalog.
#> ✔ [2025-12-25 15:37:17.386547]: Done.
#> ℹ [2025-12-25 15:37:17.388844]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:20.87149]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:20.874344]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:20.87697]: Done.
#> ℹ [2025-12-25 15:37:20.879473]: 3.498 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_17_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_17_sim.rds
#> Handling sample: Sample_19
#> ℹ [2025-12-25 15:37:21.087471]: Started.
#> ℹ [2025-12-25 15:37:21.090353]: Checking catalog.
#> ✔ [2025-12-25 15:37:21.093003]: Done.
#> ℹ [2025-12-25 15:37:21.095426]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:24.596549]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:24.599072]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:24.601451]: Done.
#> ℹ [2025-12-25 15:37:24.603854]: 3.516 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_19_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_19_sim.rds
#> Handling sample: Sample_22
#> ℹ [2025-12-25 15:37:24.804303]: Started.
#> ℹ [2025-12-25 15:37:24.807194]: Checking catalog.
#> ✔ [2025-12-25 15:37:24.809925]: Done.
#> ℹ [2025-12-25 15:37:24.812411]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:28.334829]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:28.337293]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:28.339576]: Done.
#> ℹ [2025-12-25 15:37:28.341899]: 3.538 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_22_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_22_sim.rds
#> Handling sample: Sample_24
#> ℹ [2025-12-25 15:37:28.547782]: Started.
#> ℹ [2025-12-25 15:37:28.550613]: Checking catalog.
#> ✔ [2025-12-25 15:37:28.55327]: Done.
#> ℹ [2025-12-25 15:37:28.555826]: About to start bootstrap.
#> → Bootstrapping 100 times.
#> → Total 100 times, starting no.1.
#> → Total 100 times, starting no.2.
#> → Total 100 times, starting no.3.
#> → Total 100 times, starting no.4.
#> → Total 100 times, starting no.5.
#> → Total 100 times, starting no.6.
#> → Total 100 times, starting no.7.
#> → Total 100 times, starting no.8.
#> → Total 100 times, starting no.9.
#> → Total 100 times, starting no.10.
#> → Total 100 times, starting no.11.
#> → Total 100 times, starting no.12.
#> → Total 100 times, starting no.13.
#> → Total 100 times, starting no.14.
#> → Total 100 times, starting no.15.
#> → Total 100 times, starting no.16.
#> → Total 100 times, starting no.17.
#> → Total 100 times, starting no.18.
#> → Total 100 times, starting no.19.
#> → Total 100 times, starting no.20.
#> → Total 100 times, starting no.21.
#> → Total 100 times, starting no.22.
#> → Total 100 times, starting no.23.
#> → Total 100 times, starting no.24.
#> → Total 100 times, starting no.25.
#> → Total 100 times, starting no.26.
#> → Total 100 times, starting no.27.
#> → Total 100 times, starting no.28.
#> → Total 100 times, starting no.29.
#> → Total 100 times, starting no.30.
#> → Total 100 times, starting no.31.
#> → Total 100 times, starting no.32.
#> → Total 100 times, starting no.33.
#> → Total 100 times, starting no.34.
#> → Total 100 times, starting no.35.
#> → Total 100 times, starting no.36.
#> → Total 100 times, starting no.37.
#> → Total 100 times, starting no.38.
#> → Total 100 times, starting no.39.
#> → Total 100 times, starting no.40.
#> → Total 100 times, starting no.41.
#> → Total 100 times, starting no.42.
#> → Total 100 times, starting no.43.
#> → Total 100 times, starting no.44.
#> → Total 100 times, starting no.45.
#> → Total 100 times, starting no.46.
#> → Total 100 times, starting no.47.
#> → Total 100 times, starting no.48.
#> → Total 100 times, starting no.49.
#> → Total 100 times, starting no.50.
#> → Total 100 times, starting no.51.
#> → Total 100 times, starting no.52.
#> → Total 100 times, starting no.53.
#> → Total 100 times, starting no.54.
#> → Total 100 times, starting no.55.
#> → Total 100 times, starting no.56.
#> → Total 100 times, starting no.57.
#> → Total 100 times, starting no.58.
#> → Total 100 times, starting no.59.
#> → Total 100 times, starting no.60.
#> → Total 100 times, starting no.61.
#> → Total 100 times, starting no.62.
#> → Total 100 times, starting no.63.
#> → Total 100 times, starting no.64.
#> → Total 100 times, starting no.65.
#> → Total 100 times, starting no.66.
#> → Total 100 times, starting no.67.
#> → Total 100 times, starting no.68.
#> → Total 100 times, starting no.69.
#> → Total 100 times, starting no.70.
#> → Total 100 times, starting no.71.
#> → Total 100 times, starting no.72.
#> → Total 100 times, starting no.73.
#> → Total 100 times, starting no.74.
#> → Total 100 times, starting no.75.
#> → Total 100 times, starting no.76.
#> → Total 100 times, starting no.77.
#> → Total 100 times, starting no.78.
#> → Total 100 times, starting no.79.
#> → Total 100 times, starting no.80.
#> → Total 100 times, starting no.81.
#> → Total 100 times, starting no.82.
#> → Total 100 times, starting no.83.
#> → Total 100 times, starting no.84.
#> → Total 100 times, starting no.85.
#> → Total 100 times, starting no.86.
#> → Total 100 times, starting no.87.
#> → Total 100 times, starting no.88.
#> → Total 100 times, starting no.89.
#> → Total 100 times, starting no.90.
#> → Total 100 times, starting no.91.
#> → Total 100 times, starting no.92.
#> → Total 100 times, starting no.93.
#> → Total 100 times, starting no.94.
#> → Total 100 times, starting no.95.
#> → Total 100 times, starting no.96.
#> → Total 100 times, starting no.97.
#> → Total 100 times, starting no.98.
#> → Total 100 times, starting no.99.
#> → Total 100 times, starting no.100.
#> Bootstrap done.
#>
#> ✔ [2025-12-25 15:37:32.150843]: Signature exposures collected.
#> ✔ [2025-12-25 15:37:32.153289]: Errors and similarity collected.
#> ✔ [2025-12-25 15:37:32.155722]: Done.
#> ℹ [2025-12-25 15:37:32.158028]: 3.61 secs elapsed.
#> Save expo result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_24_expo.rds
#> Save similarity result to temp file /tmp/RtmpKIbCyr/sigminer_attribute_activity/Sample_24_sim.rds
#>
#> Reading result files...
#> Clean cache files when exits.
#> Done.
expo$abs_activity
#> sample Sig1 Sig2 Sig3 Sig4 Sig5
#> <char> <num> <num> <num> <num> <num>
#> 1: Sample_2 0.0000 8324.3129 0.00000 0.00000 836.23229
#> 2: Sample_3 0.0000 5134.2448 0.00000 0.00000 1711.02871
#> 3: Sample_4 0.0000 7628.7041 1523.89284 0.00000 0.00000
#> 4: Sample_5 0.0000 1033.6448 0.00000 0.00000 0.00000
#> 5: Sample_6 0.0000 0.0000 0.00000 0.00000 181.69335
#> 6: Sample_7 0.0000 582.8461 0.00000 0.00000 172.01602
#> 7: Sample_8 0.0000 0.0000 0.00000 0.00000 1369.45498
#> 8: Sample_9 0.0000 0.0000 0.00000 0.00000 0.00000
#> 9: Sample_10 1143.1114 0.0000 0.00000 0.00000 0.00000
#> 10: Sample_11 0.0000 1432.9232 0.00000 0.00000 1600.13559
#> 11: Sample_13 0.0000 1570.0646 0.00000 0.00000 56.91541
#> 12: Sample_14 0.0000 1331.7105 0.00000 0.00000 0.00000
#> 13: Sample_15 0.0000 3713.2011 2380.78415 0.00000 551.04363
#> 14: Sample_16 673.5523 1095.9283 0.00000 2397.98433 0.00000
#> 15: Sample_18 0.0000 0.0000 0.00000 0.00000 178.07434
#> 16: Sample_20 440.5282 0.0000 0.00000 822.08119 416.87212
#> 17: Sample_21 0.0000 3630.3894 4917.63789 0.00000 0.00000
#> 18: Sample_23 0.0000 0.0000 0.00000 33.20335 243.22641
#> 19: Sample_25 0.0000 0.0000 0.00000 0.00000 2141.32871
#> 20: Sample_26 0.0000 429.4069 0.00000 0.00000 2491.68582
#> 21: Sample_27 0.0000 6699.1632 76.18949 0.00000 194.17338
#> 22: Sample_28 302.2635 0.0000 0.00000 818.94743 0.00000
#> 23: Sample_29 0.0000 0.0000 0.00000 1097.81229 578.42602
#> 24: Sample_30 400.9180 0.0000 0.00000 826.83399 238.78021
#> 25: Sample_1 9138.4848 0.0000 0.00000 18252.04649 0.00000
#> 26: Sample_12 8847.7116 2860.9798 5597.61520 18490.61967 0.00000
#> 27: Sample_17 8282.3148 0.0000 0.00000 0.00000 12385.06731
#> 28: Sample_19 10367.8524 1558.8368 0.00000 4600.13526 5734.80507
#> 29: Sample_22 3572.9161 0.0000 0.00000 11095.51018 8944.23850
#> 30: Sample_24 0.0000 15548.7912 22224.83696 0.00000 0.00000
#> sample Sig1 Sig2 Sig3 Sig4 Sig5
#> <char> <num> <num> <num> <num> <num>
#> Sig6 Sig7 Sig8 Sig9 Sig10 Sig11
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.00000 0.0000 0.0000 0.00000 0.0000 9633.1075
#> 2: 0.00000 199.5056 0.0000 247.33530 3412.1065 0.0000
#> 3: 0.00000 1005.7703 0.0000 330.72220 0.0000 135.6430
#> 4: 0.00000 0.0000 0.0000 0.00000 0.0000 1661.3552
#> 5: 0.00000 226.9018 0.0000 0.00000 0.0000 1197.6510
#> 6: 0.00000 307.6999 0.0000 367.55856 149.6321 0.0000
#> 7: 0.00000 482.2529 0.0000 0.00000 0.0000 537.6053
#> 8: 0.00000 0.0000 2783.9792 5340.02083 0.0000 0.0000
#> 9: 2541.17025 1129.0811 1701.7448 0.00000 0.0000 3136.6450
#> 10: 0.00000 0.0000 0.0000 81.98904 192.4109 1940.1209
#> 11: 13.19321 401.8427 0.0000 46.59928 0.0000 0.0000
#> 12: 0.00000 0.0000 0.0000 0.00000 0.0000 2879.2895
#> 13: 0.00000 0.0000 0.0000 3589.13531 0.0000 524.3026
#> 14: 430.87300 2892.9436 0.0000 0.00000 238.9572 0.0000
#> 15: 405.92264 0.0000 2134.3934 2835.14250 0.0000 0.0000
#> 16: 40.38733 728.0084 0.0000 0.00000 148.8330 0.0000
#> 17: 3540.94597 502.9650 0.0000 561.13782 0.0000 0.0000
#> 18: 532.02063 1582.7356 0.0000 847.56904 1161.9916 0.0000
#> 19: 0.00000 2796.0676 1904.0547 2645.43897 0.0000 698.4211
#> 20: 0.00000 0.0000 0.0000 926.44717 2092.5266 0.0000
#> 21: 1587.79622 906.3852 0.0000 212.54317 0.0000 0.0000
#> 22: 362.25152 1080.0412 0.0000 0.00000 402.3160 0.0000
#> 23: 176.34483 0.0000 965.4548 830.89664 0.0000 0.0000
#> 24: 253.43957 0.0000 223.5043 100.89195 882.4947 0.0000
#> 25: 13782.50908 0.0000 0.0000 0.00000 1493.6083 0.0000
#> 26: 3816.65478 9141.6032 0.0000 0.00000 1195.2922 0.0000
#> 27: 0.00000 0.0000 1629.9891 0.00000 2218.2940 3163.1918
#> 28: 1724.13326 7400.3912 0.0000 0.00000 1469.2849 0.0000
#> 29: 5109.09700 0.0000 3655.6147 0.00000 6016.8222 0.0000
#> 30: 679.65994 5732.9665 0.0000 1757.38556 0.0000 0.0000
#> Sig6 Sig7 Sig8 Sig9 Sig10 Sig11
#> <num> <num> <num> <num> <num> <num>
# }
if (FALSE) { # \dontrun{
# Iterative extraction:
# This procedure will rerun extraction step
# for those samples with reconstructed catalog similarity
# lower than a threshold (default is 0.95)
e2 <- bp_extract_signatures_iter(
t(simulated_catalogs$set1),
range = 9:11,
n_bootstrap = 5,
n_nmf_run = 5,
sim_threshold = 0.99
)
e2
# When the procedure run multiple rounds
# you can cluster the signatures from different rounds by
# the following command
# bp_cluster_iter_list(e2)
## Extra utilities
rank_score <- bp_get_rank_score(e1)
rank_score
stats <- bp_get_stats(e2$iter1)
# Get the mean reconstructed similarity
1 - stats$stats_sample$cosine_distance_mean
} # }