Sigminer: A Scalable Toolkit to Extract, Analyze and Visualize Mutational Signatures
last revised on 2021-05-18
Chapter 1 Introduction
Underlying cancer hallmarks are genome instability, which generates the genetic diversity that expedites their acquisition, and inflammation, which fosters multiple hallmark functions (Hanahan 2011). Cancer genomes typically harbors more than 1,000 mutations in small (e.g., point mutations, short insertions and deletions) and large scale (e.g., copy number variations, rearrangements). Genomic contexts where mutation may accumulate in response to both endogenous processes and exogeneous exposures. In recent years, computational approaches (typically non-negative matrix factorization (NMF)) have been applied to the mutation catalog analysis of human/mouse tumors to detect characteristic mutational patterns, also known as “mutational signatures”.
1.1 Biological Significance of Mutational Signature
To illustrate the biological significance of mutational signatures, we show some well organized figures here.
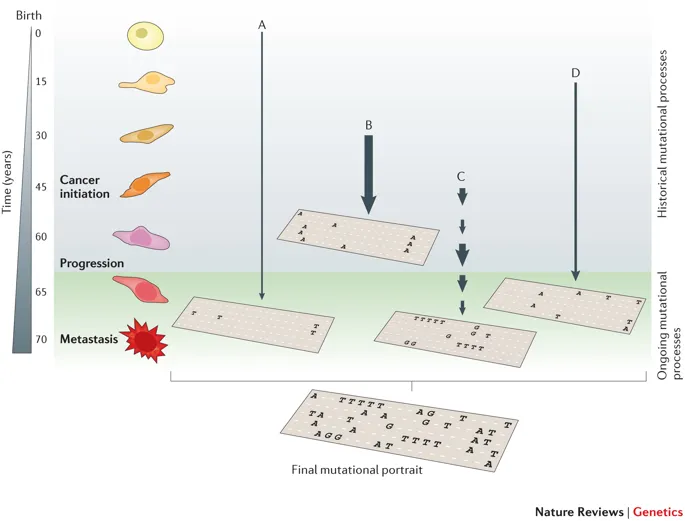
Figure 1.1: The illustration of SBS signature, fig source: https://www.nature.com/articles/nrg3729
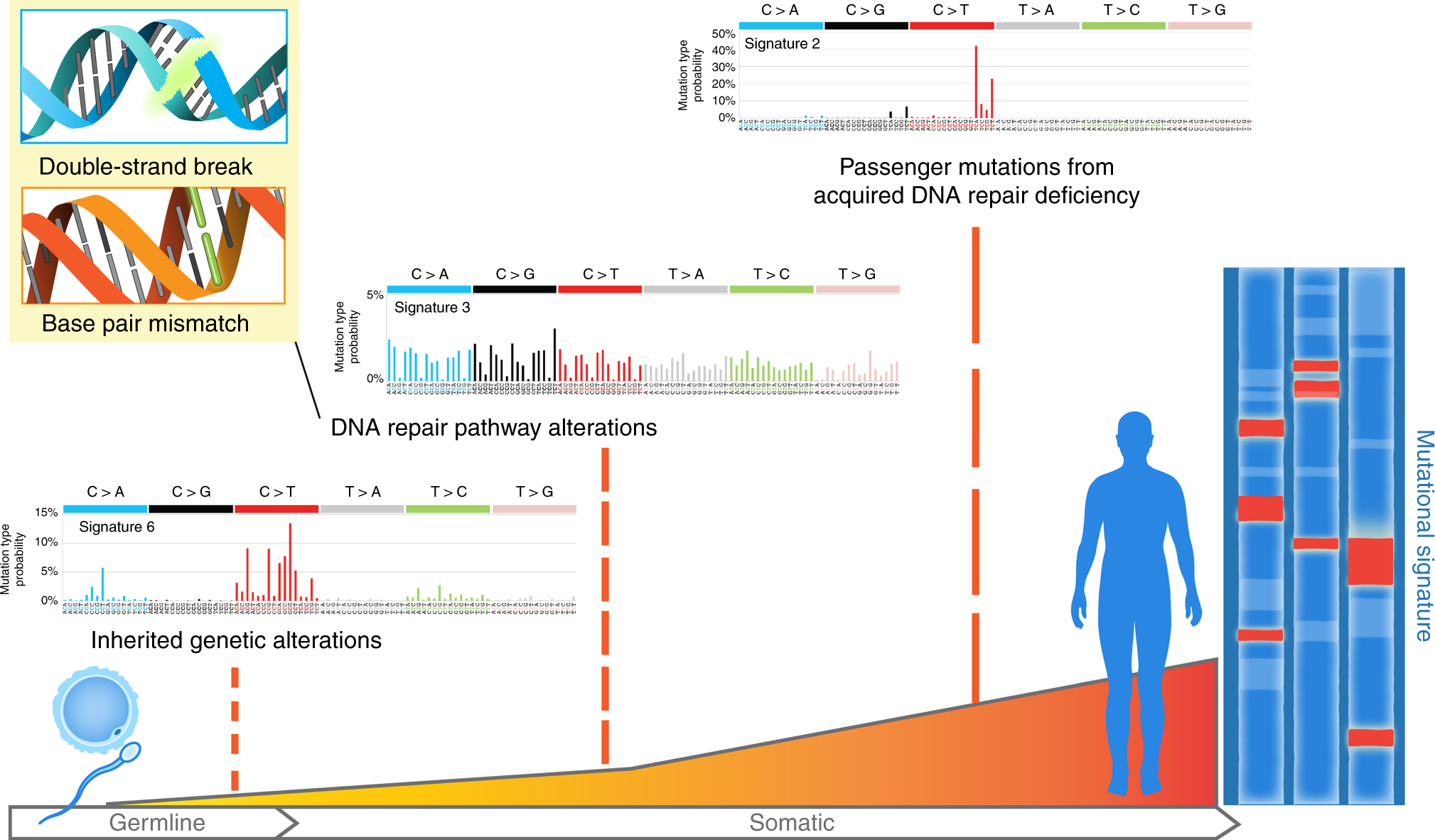
Figure 1.2: The illustration of SBS signature (2), fig source: https://www.nature.com/articles/s41467-018-05228-y
SBS (short for single base substitution) signature is a famous type of mutational signature. SBS signatures are well studied and related to single-strand changes, typically caused by defective DNA repair. Common etiologies contain aging, defective DNA mismatch repair, smoking, ultraviolet light exposure and APOBEC.
Currently, all SBS signatures are summarized in COSMIC database, including two versions: v2 and v3.
Recently, Alexandrov et al. (2020) extends the concept of mutational signature to three types of alteration: SBS, DBS (short for doublet base substitution) and INDEL (short for short insertion and deletion). All reported common signatures are recorded in COSMIC (https://cancer.sanger.ac.uk/cosmic/signatures/), so we usually call them COSMIC signatures.
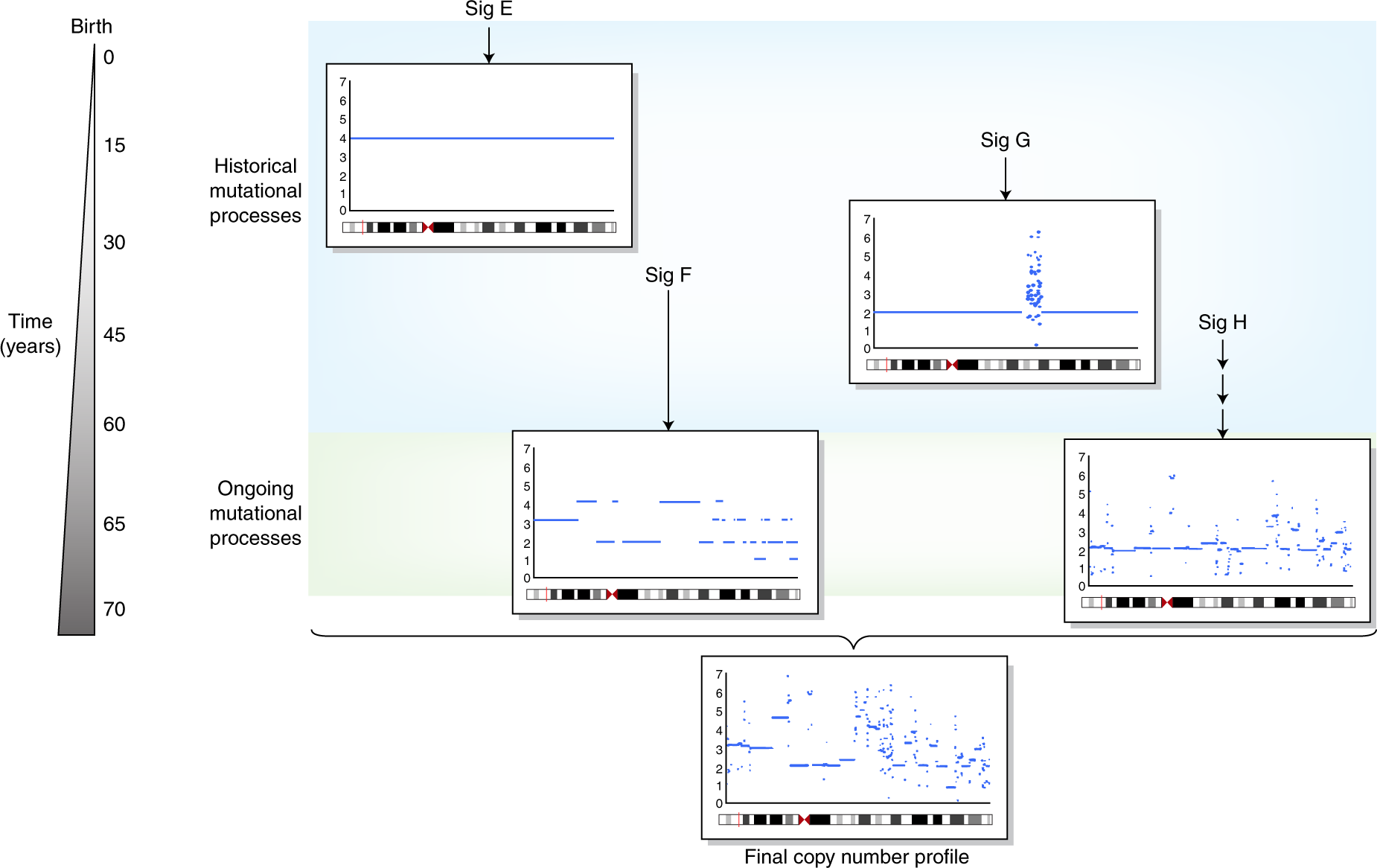
Figure 1.3: The illustration of copy number signatures, fig source: https://www.nature.com/articles/s41588-018-0212-y
Copy number signatures are less studied and many works are still to be done. The introduction is described in Chapter 3.
Genome rearrangement signatures are limited to whole genome sequencing data and also less studied, the implementation is not available in current version of Sigminer. We are happy to accept a PR if you are interested in create an extension function to Sigminer.
More details about mutational signatures you can read the wiki page.
1.2 Sigminer
Here, we present an easy-to-use and scalable toolkit for mutational signature analysis and visualization in R. We named it sigminer (signature + miner).
This tool can help users to extract, analyze and visualize signatures from genomic alteration records,
thus providing new insight into cancer study.
1.3 Installation
The stable release version of sigminer package can be installed from the CRAN:
install.packages("sigminer", dependencies = TRUE)
# Or
BiocManager::install("sigminer", dependencies = TRUE)Set
dependencies = TRUEis recommended because many packages are required for full features in sigminer.
The development version of sigminer package can be installed from Github:
# install.packages("remotes")
remotes::install_github("ShixiangWang/sigminer", dependencies = TRUE)1.4 Issues or Suggestions
Any issue or suggestion can be posted on GitHub issue, we will reply ASAP.
Any pull requrest is welcome.
1.5 Preparation
To reproduce the examples shown in this manual, users should load the following packages firstly. sigminer is requred to have version >= 1.0.0.
library(sigminer)
#> sigminer version 2.0.0
#> - Star me at https://github.com/ShixiangWang/sigminer
#> - Run hello() to see usage and citation.
library(NMF)
#> Loading required package: pkgmaker
#> Loading required package: registry
#> Loading required package: rngtools
#> Loading required package: cluster
#> NMF - BioConductor layer [OK] | Shared memory capabilities [NO: bigmemory] | Cores 7/8
#> To enable shared memory capabilities, try: install.extras('
#> NMF
#> ')Current manual uses sigminer 2.0.0. More info about sigminer can be given as:
hello()
#> Thanks for using 'sigminer' package!
#> =========================================================================
#> Version: 2.0.0
#> Run citation('sigminer') to see how to cite sigminer in publications.
#>
#> Project home : https://github.com/ShixiangWang/sigminer
#> Bug report : https://github.com/ShixiangWang/sigminer/issues
#> Documentation: https://shixiangwang.github.io/sigminer-doc/
#> =========================================================================
#> 1.6 Overview of Contents
The contents of this manual have been divided into 4 sections:
- Common workflow.
- de novo signature discovery.
- single sample exposure quantification.
- subtype prediction.
- Target visualization.
- copy number profile.
- copy number distribution.
- catalogue profile.
- signature profile.
- exposure profile.
- …
- Universal analysis.
- association analysis.
- group analysis.
- Other utilities.
All functions are well organized and documented at https://shixiangwang.github.io/sigminer/reference/index.html (For Chinese users, you can also read it at https://shixiangwang.gitee.io/sigminer/reference/index.html). For usage of a specific function fun, run ?fun in your R console to see its documentation.
1.7 Citation and LICENSE
If you use sigminer in academic field, please cite one of the following papers.
Wang S, Li H, Song M, Tao Z, Wu T, He Z, et al. (2021) Copy number signature analysis tool and its application in prostate cancer reveals distinct mutational processes and clinical outcomes. PLoS Genet 17(5): e1009557. https://doi.org/10.1371/journal.pgen.1009557
Shixiang Wang, Ziyu Tao, Tao Wu, Xue-Song Liu, Sigflow: An Automated And Comprehensive Pipeline For Cancer Genome Mutational Signature Analysis, Bioinformatics, btaa895. https://doi.org/10.1093/bioinformatics/btaa895
The software is made available for non commercial research purposes only under the MIT. However, notwithstanding any provision of the MIT License, the software currently may not be used for commercial purposes without explicit written permission after contacting Shixiang Wang wangshx@shanghaitech.edu.cn or Xue-Song Liu liuxs@shanghaitech.edu.cn.
MIT © 2019-2020 Shixiang Wang, Xue-Song Liu
MIT © 2018 Anand Mayakonda
Cancer Biology Group @ShanghaiTech
Research group led by Xue-Song Liu in ShanghaiTech University